
ゲノムワイド関連解析(ゲノムワイドかんれんかいせき、英: genome-wide association study, GWA study、略称: GWAS)、またはゲノムワイド関連研究は、ゲノミクス(ゲノム科学)において、異なる個人のゲノム全域にわたる(ゲノムワイドな)遺伝的変異一式を対象に、ある形質に関連する変異があるかどうかを調べる観察研究である。GWAS は、通常、一塩基多型(SNP)とヒトの主要な疾患などの形質との関連に焦点を当てているが、他のすべての遺伝的変異や他の生物にも同様に適用することができる。
概要
GWASをヒトのデータに適用した場合、特定の形質や疾患について様々な表現型を持つ参加者のDNAを比較する。これらの参加者は、疾患のある人(症例)と疾患のない同様の人(対照)であったり、血圧などの特定の形質について異なる表現型を持つ人であったりする。この方法は、遺伝子型を優先するのではなく、臨床症状によって参加者を分類する表現型優先アプローチとして知られている。一人一人のDNAを採取し、そこからSNPアレイを用いて読み取る。ある種の遺伝子変異(1つの対立遺伝子)が病気の人に多く見られる場合、その遺伝子変異体はその病気と関連していると言われる。そして、関連するSNPは、疾患のリスクに影響を与える可能性のあるヒトゲノムの領域を示していると考えられる。
GWASは、あらかじめ指定された少数の遺伝子領域を特異的に検証する方法とは対照的に、ゲノム全体を調査する。したがって、GWASは、遺伝子特異的な仮説駆動型の研究とは対照的に、データ駆動型の研究といえる。 GWASでは、疾患に関連するDNAのSNPやその他のバリアントを特定するが、それだけではどの遺伝子が原因であるかを特定することはできない。
2002年に発表された最初のGWASは、心筋梗塞を対象としたものだった。その後、この研究デザインは2005年に、加齢黄斑変性の患者を調査した画期的なGWASに導入され、健康対照者と比較して対立遺伝子の頻度が有意に変化した 2つのSNPが発見された。2017年現在、ヒトを対象とした3,000件以上のGWASで、1,800以上の疾患や形質が調査され、何千ものSNPの関連が発見された。希少な遺伝子疾患の場合を除き、これらの関連性は非常に弱くリスクの多くを説明できないかもしれないが、重要かもしれない遺伝子や経路についての知見を得ることができる。
背景
ヒトゲノムは、2つあれば何百万通りもの違いがある。ゲノムの個々のヌクレオチドには小さな変異(SNP)があり、欠失、挿入、コピー数の変異などの大きな変異もある。これらの変異は、病気のリスクから身長などの身体的特徴まで、個人の形質(表現型)に変化をもたらす可能性がある。2000年頃、GWASが導入される前の主な調査方法は、家族の遺伝的連鎖を調べる遺伝学的調査(連鎖研究)だった。この方法は、単一遺伝子疾患に対しては非常に有用であることがわかっていたが、一般的な疾患や複雑な疾患では、連鎖研究の調査結果を再現することは困難だった 。そこで連鎖研究に変わるものとして提案されたのが、遺伝子関連研究だった。この研究は、ある遺伝子変異の対立遺伝子が、対象となる表現型(例: 研究対象となる疾患)を持つ個人に予想以上に多く見られるかどうかを調べるものである。初期の統計的検出力の計算で、弱い遺伝的影響を検出するには、連鎖研究よりもこの方法の方が優れていることが示された。
概念的な枠組みに加えて、GWASを可能にした要因がいくつかある。1つは、バイオバンクの登場であった。バイオバンクとは、ヒトの遺伝物質を保管する機関で、研究に必要な数の生物学的標本を集める費用と難易度を大幅に下げてくれた。もう1つは、2003年から始まった国際HapMap計画によって、GWASで調べられる一般的なSNPの大部分が特定されたことであった。国際HapMap計画で特定されたハプロブロック構造は、変動の大部分を記述するSNPのサブセットに焦点を当てることを可能にした。また、ジェノタイピングアレイを用いてこれらのSNPをすべてジェノタイピングする方法を開発することも重要な前提条件だった。
方法
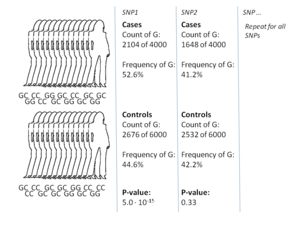
GWASの最も一般的なアプローチは、症例対照研究で、2つの大規模な個人グループ、すなわち健康な対照グループと疾患に罹患した症例グループを比較する。各グループのすべての個人は、一般的な既知のSNPの大部分について遺伝子型別を解析される。SNPの正確な数はジェノタイピング技術によって異なるが、通常は100万個以上である。そして、これらのSNPsのそれぞれについて、対立遺伝子の頻度が症例群と対照群の間で有意に変化しているかどうかを調べる。このような設定では、効果の大きさを報告するための基本的な単位はオッズ比である。オッズ比とは、2つのオッズの比であり、GWASの文脈では、特定の対立遺伝子を持つ個人の症例のオッズと、同じ対立遺伝子を持たない個人の症例のオッズを指す。
例として、TとCの 2つの対立遺伝子があるとする。対立遺伝子Tを持つ症例群の個体数は「A」、対立遺伝子Tを持つ対照群の個体数は「B」で表される 。同様に、対立遺伝子Cを有する症例群の個体数は「X」、対立遺伝子Cを有する対照群の個体数は「Y」で表される。この場合、対立遺伝子Tのオッズ比はA:B(標準的な用語ではBに対するAの比)をX:Yで割ったものであり、数学表記では単純に(A / B)/(X / Y)となる。
症例群の対立遺伝子の頻度が対照群よりもはるかに高い場合、オッズ比は1よりも高くなり、対立遺伝子頻度が低い場合はその逆となる。さらに、オッズ比の有意性を示す P値は、通常、単純なカイ二乗検定を用いて算出される。1とは有意に異なるオッズ比を見つけることは、SNPが疾患と関連していることを示すため、GWASの目的となる。非常に多くのバリアントが検定されるため、p値が 5×10−8 未満であることをもって有意であると判断することが標準的である。
この症例対照アプローチにはいくつかの変法がある。症例対照GWASの一般的な代替法は、定量的な表現型データ、例えば身長やバイオマーカーの濃度、さらには遺伝子発現などの分析である。同様に、優性または劣性の浸透度パターン用に設計された代替統計を使用することができる。SNPTESTやPLINKなどのバイオインフォマティクス・ソフトウェアを用いて計算するのが一般的で、これらの代替統計の多くに対応している。GWASでは、個々のSNPの影響に注目する。しかし、複数のSNP間の複雑な相互作用(エピスタシス)が、複雑な疾患の原因となっている可能性もある。相互作用の数は指数関数的に増加する可能性があるため、GWASデータから統計的に有意な相互作用を検出することは、計算的にも統計的にも困難である。この課題は、データマイニングから着想を得たアルゴリズムを使用する既存の出版物で取り組まれていきた。さらに、研究者たちは、GWASのデータをタンパク質間相互作用ネットワークなどの他の生物学的データと統合して、より有益な結果を引きだそうとしている。
大半のGWASでは、研究に使用された遺伝子型チップにないSNPの遺伝子型を代入することが重要なステップとなっている。このプロセスにより、関連性を検証できるSNPの数が大幅に増え、研究の検出力が向上し、異なるコホート間のGWASのメタ分析が容易になる。遺伝子型の代入は、GWASデータとハプロタイプのリファレンス・パネルとを組み合わせた統計的手法によって行われる。これらの方法では、短い配列の間の個人間でハプロタイプが共有されていることを利用して、対立遺伝子を推定する。ジェノタイプ・インピュテーションのための既存のソフトウェア・パッケージには、IMPUTE2、Minimac、Beagle、MaCHなどがある 。
関連性の算出に加えて、結果を交絡させる可能性のある因子を考慮するのが一般的である。性別と年齢は交絡因子の一般的な例である。さらに、多くの遺伝的変異は、その変異が最初に生じた地理的・歴史的集団と関連していることも知られている。このような関連性があるため、研究では、参加者の地理期的・民族的背景を考慮して、いわゆる母集団の層別化を行う必要がある。そうしないと、これらの研究は偽陽性の結果を生み出す可能性がある。
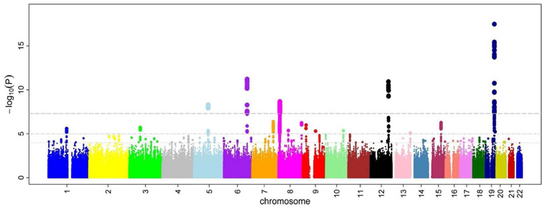
すべてのSNPについてオッズ比とP値を計算した後で、マンハッタン・プロットを作成するのが一般的である。GWASでは、このプロットはP値の負の対数をゲノム位置の関数として表している。このようにして、最も有意な関連性を持つSNPは、ハプロブロック構造のため、通常、点のスタックとしてプロット上に表示される。重要なのは、有意性を示すP値のしきい値が、多重検定の問題に対して補正されていることである。閾値の正確な値は研究によって異なるが 、数十万から数百万の検定においても有意であることを示す 5×10−8が用いられてきた。GWASでは通常、最初の解析を発見コホートで行い、その後、独立した検証コホートで最も有意なSNPを検証する。
結果

GWASから同定されたSNPの包括的なカタログを作成する試みがなされている。2009年の時点で、疾患に関連するSNPは数千にのぼる。
2005年に実施された最初のGWASでは、96人の加齢黄斑変性(ARMD)患者と50人の健康対照者を比較した 。その結果、両群間で対立遺伝子の頻度が有意に変化している 2つのSNPを同定した。これらのSNPは、補体因子Hをコードする遺伝子に位置しており、これはARMDの研究では予想外の発見だった。この最初のGWASで得られた知見は、その後、ARMDにおける補体系の治療的操作に向けて、さらなる機能的研究を促した。GWASの歴史におけるもう1つの画期的な発表はウェルカム・トラスト・ケース・コントロール・コンソーシアム(WTCCC)研究であり、2007年に発表された時点では最大のGWASだった。WTCCCでは、7つの一般的な疾患の14,000症例(冠状動脈性心臓病、1型糖尿病、2型糖尿病、関節リウマチ、クローン病、双極性障害、高血圧のそれぞれについて約2,000人)と、3,000人の共通対照群が含まれていた。この研究は、これらの疾患の基礎となる多くの新しい病気の遺伝子を発見することに成功した。
これらの最初の画期的なGWAS以来、2つの一般的な傾向がある。1つは、より大きなサンプルサイズへと向かっている。2018年には、学歴に関するもので110万人、不眠症に関するもので130万人と、100万人以上のサンプルサイズに達するGWASがある。その理由は、オッズ比が小さく、対立遺伝子頻度が低いリスクSNPを確実に検出しようという動きがあるからである。もう1つの傾向は、血中脂質やプロインスリンなどのバイオマーカーのように、より狭く定義された表現型を使用することである。これらは中間表現型と呼ばれ、その解析はバイオマーカーの機能的研究に役立つ可能性がある。GWASの変法では、疾患を持つ人の一親等の親族を対照にする。このタイプの研究は、代理人 (proxy) によるゲノムワイド関連研究(GWAX)と呼ばれる。
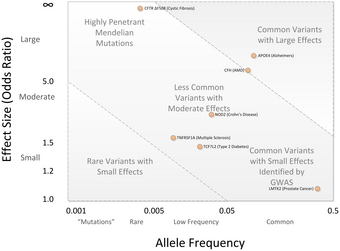
GWASに関する議論の中心は、GWASによって発見されたSNP変異のほとんどが、わずかな疾患のリスクの増加にしか関連しておらず、小さな予測値しか持たないということだった。オッズ比の中央値は、一つのリスクSNPあたり1.33であり、3.0を超えるオッズ比を示すのはわずかである。オッズ比の大きさは小さく、遺伝性変動をほとんど説明できていない。この遺伝性変動は、一卵性双生児を対照とした遺伝率調査から推定される。例えば、身長の変動の80 - 90%は遺伝的差異によって説明できることが知られているが、GWASでは、この変動のごく一部しか説明できていない。
関連項目
外部リンク
- omicX上の遺伝子型-表現型相互作用ソフトウェアツールおよびデータベース
- ゲノムワイド関連研究の分析のための統計的方法[ビデオ講義シリーズ]
- 全ゲノム関連研究—国立ヒトゲノム研究所による
- GWAS Central —要約レベルの遺伝的関連の調査結果の中央データベース
- Barrett (2010年7月18日). “How to read a genome-wide association study”. Genomes Unzipped. 2021年8月4日閲覧。
- ゲノムワイド関連研究のコンソーシアム(GWAS) — Bennett SN、Caporaso、NEなどによる。
- PLINK —全ゲノム関連解析ツールセット
- ENCODEスレッドエクスプローラーバリエーションの理解に対する機能情報の影響。ネイチャー(ジャーナル)