
| 原発性線毛運動不全症 | |
|---|---|
| 別称 | 線毛機能不全症候群 |

| |
| 運動性繊毛9+2構造(A) ダイニン腕欠損(B) | |
原発性線毛運動不全症( 線毛機能不全症候群 primary ciliary dyskinesia; PCD )は、気管・気管支、鼻副鼻腔、耳管、中耳、卵管などを覆う繊毛の運動に異常をきたす稀な遺伝性疾患であり、多くは常染色体潜性の遺伝形式を示す。気道上皮細胞の表面に見られる運動性繊毛は、有効打と回復打の反復によって粘液を外へ向かって移動させる複雑な構造を持つ細胞小器官である。繊毛運動が障害されると病原体の排除に有効な粘液の排出能力が低下して上・下気道の感染症が生じやすくなる。
同一疾患について原発性線毛機能不全症の訳語も用いられている。有効な協調運動の繰り返しがなければ繊毛は動いていても動かない場合と同様な病態を呈することから、線毛不動症候群(immotile cilia syndrome)という旧来の用語は使われなくなっている。cilium(複数形cilia)は、「繊毛」と表記されるが、医学用語としては「線毛」が用いられている。
臨床像
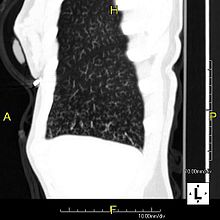


PCDの臨床像は、日本で用いられている副鼻腔気管支症候群(sinobronchial syndrome; SBS)の概念に合致しており、上・下気道症状から本症以外の原因による気管支拡張症と鑑別することは困難である。特にびまん性汎細気管支炎(diffuse panbronchiolitis; DPB)のCT画像上の特徴とされる小葉中心性のびまん性粒状陰影が認められる場合は鑑別に苦慮する。PCDでは気道系からの粘液排出が障害されることで、副鼻腔炎、気管支炎、肺炎、中耳炎などが慢性化、再発しやすい。感染症の予防と制御を早期に開始することが重要である。
原因不明の新生児呼吸困難、生後早期に発症する鼻炎、幼少期に始まる進行性の気管支拡張症に伴う咳、痰などは本症だけに見られる特徴ではないため、診断が遅れがちであり、進行すると重症例では肺移植が必要となる場合がある。小児期に難聴など滲出性中耳炎の遷延に伴う症状が見られることも多い。副鼻腔内の粘液貯留のため嗅覚異常も認められる。男性の場合、原因遺伝子によって精子の運動機能も障害され、しばしば不妊症が見られるが、顕微受精などにより妊娠率は向上している。 また卵管繊毛の運動障害による不妊も報告されている。
また原因遺伝子によっては、後述のように内臓逆位が約半数に認められる。また稀に先天性の水頭症が認められる。一般に本疾患の発症率は1万人から2万人の出生に1人と推定されてきたが、国や地域により原因となる遺伝子頻度が著しく異なり、特にアジア・アフリカ諸国ではいまだ確定診断に至る例が限られているため、発症率は過小評価されているものと考えられる。
遺伝子異常
PCDは、多数のタンパク質から構成される運動性繊毛の構造や機能が障害される遺伝的に不均一な疾患である 。繊毛を正しく動かすのに必要な構造として、ダイニン外腕、ダイニン内腕、ネキシン−ダイニン制御複合体、放射状スポーク、中心微小管構造が知られており、いずれの障害によっても上・下気道症状が認められる。繊毛の発生、維持に関与する遺伝子も含めて、2022年現在までに約50個の原因遺伝子が特定されている。主に常染色体潜性の遺伝形式を示すため、ほとんどの原因遺伝子では両アレルに病的異常があって初めてPCDの症状、所見を呈する。例えば、DNAH5遺伝子の両アレルの異常は、欧米で高頻度に認められ、超微細構造上、ダイニン外腕欠損を生じる。
日本では、ネキシン−ダイニン制御複合体を構成するDRC1(CCDC164)遺伝子のエクソン1−4を包含する27,748 bpに及ぶ大規模欠失アレルが、一般集団の0.2%程度(0.0-0.7%)に見出され、そのホモ接合体がPCDの原因として最も大きな割合を占めるものと推定されている。この遺伝子異常では鼻腔内一酸化窒素(nitric oxide; NO)が低値であるが、内臓逆位は見られず、電子顕微鏡像に目立った異常が見られないことが知られている。マルチプレックスPCR法によって欠失アレルの有無を検出することが可能である。
病態生理

遺伝子の先天的な異常により、繊毛を構成する細長い構造体である軸糸の運動が胎児期より障害され、生後、上・下気道の感染症が生じやすくなる。
発生初期に中心対微小管を持たないノード繊毛と呼ばれる特殊な繊毛が原始結節に出現し、自律的に回転運動を行うことで左向きの流れが生じ、臓器の左右非対称性が生み出される、軸糸異常の中で、特にダイニン腕の欠損を生じると、このノード繊毛の運動も障害を受けて左右の決定ができなくなり、約50%の確率で内臓逆位が生じることになる。また一部の症例では、多脾症、無脾症、複雑な心奇形などを伴う内臓錯位が見られる。内臓逆位、慢性副鼻腔炎、気管支拡張症の3徴候が見られる時、歴史的に本症はKartagener症候群と呼ばれてきた。
また運動性繊毛の異常によるPCD と非運動性の一次繊毛の異常に起因するその他の遺伝性疾患(多発性嚢胞腎、 Bardet-Biedl 症候群、Meckel-Gruber 症候群など)を繊毛病 ciliopathy と総称することがある。

診断
欧州では臨床像からPCDの可能性を予測する、PICADARスコアが広く利用されている。本症の診断にはいくつかの検査法が開発されている。スクリーニング検査として鼻腔内NO産生量測定、繊毛の打数と打ち方を調べる高速ビデオ顕微鏡検査がある。繊毛運動に関わる主要タンパク質の免疫染色も行われている。疑い例には電子顕微鏡検査による繊毛横断面の微細形態の観察が行われる。近年、微細形態に明らかな異常を認めず、内臓逆位も生じないが、PCDの原因となりうる遺伝子異常が次々と見出されている。昨今の遺伝子配列決定技術の著しい進歩に伴い、原因遺伝子の病的バリアント同定にはターゲット・リシークエンシングやエクソーム・シークエンシングが積極的に活用されているが、2022年現在も遺伝子検査によって確定診断に至る割合は70−75%にとどまっている。
遺伝子検査は遺伝カウンセリングが提供できる体制で実施する必要がある。伝統的に高速ビデオ顕微鏡所見を重視する欧州と、重視しない米国では異なる診断アルゴリズムが採用されており、現時点では統合されていない。かつて用いられてきたサッカリンテストは精度面で問題があり、現在は推奨されない。

治療
PCDの治療法は臓器別、対症的に嚢胞性線維症(cystic fibrosis; CF)やそれ以外の原因による気管支拡張症に準じて行われており、稀少疾患である本症のみを対象とした臨床治験は乏しいのが現状である。胸部理学療法により喀痰の排出を促し、ワクチン等で呼吸器系の感染症を予防し、適切な抗菌薬により急性増悪を抑えるべきである。禁煙、運動療法も重要である。マクロライド系抗菌薬による長期治療が経験的に行われることが多いが、急性増悪の頻度を減少させる以外の効果は十分でないとされる。
日本で「線毛機能不全症候群(カルタゲナー(Kartagener)症候群を含む。)」は、小児慢性特定疾病のひとつとして医療費助成の対象となっているが、成人では2023年3月の厚生科学審議会 (疾病対策部会指定難病検討委員会)において、同疾患を指定難病に追加指定することが了承された。また日本鼻科学会より「線毛機能不全症候群の診療の手引き」が刊行されている。
予後
PCD患者の平均余命については十分なデータが得られてない。海外の多施設研究によると本症の肺機能は嚢胞性線維症より一般に良好であるが、加齢とともに緑膿菌の定着、気流制限(1秒率の低下)が進行することが多い。また原因遺伝子の種類により重症度に差が見られることが報告されている。
歴史
内臓逆位と気管支拡張の合併例については、1904年にAlfons Siewertによって最初に記述されていたが 、1933年にManes Kartagenerは副鼻腔炎、気管支拡張症、内臓逆位の3徴候を示す4人の患者を報告し、その後も数十年にわたって家族発症例を含む同様の症例を集積した。一方、1970年代にストックホルムの超微細構造学者であったBjorn Afzeliusらは、男性不妊症で動きの見られない精子に着目し、研究を進めた。当時、精子鞭毛軸糸の微細形態は、呼吸上皮の運動性繊毛の構造と酷似していることが知られており、さらに電子顕微鏡下で観察される軸糸ダイニン腕の欠損と粘液繊毛輸送能の低下が明らかな症例に、上記の3徴候(カルタゲナーの三徴)が確認されたため、鞭毛、繊毛軸糸の運動異常と男性不妊、慢性気道感染症、臓器の左右非対称性との関連性についての報告を行った。それ以来、現在に至るまでこの分野の研究は大きく発展している。
脚注
参考文献
外部リンク
- The Genetic Disorders of Mucociliary Clearance Consortium
- PCD Foundation
- BEAT-PCD ; Better Experimental Approaches to Treat PCD
| 分類 | |
|---|---|
| 外部リソース(外部リンクは英語) |