
進行性骨化性線維異形成症(しんこうせい こつかせい せんいいけいせいしょう、英: Fibrodysplasia Ossificans Progressiva、頭字語:FOP)は、全身の筋肉や腱が骨に変化してしまう病気。結合組織に発生する稀な遺伝子疾患である。身体の矯正メカニズムが線維性組織に起こす難病であり、筋肉・腱・靭帯が骨組織に変化して硬化する。多くの症例において、それらの症状によって関節が固定されて動かなくなる。外科的治療は、手術自体の侵襲によって周囲組織の骨化が促進されることになるために、通常行うことができない。
発症率は200万人に1人といわれている。2022年時点で日本には約80人の患者がいる。
2007年3月12日に開かれた厚生労働省の第4回特定疾患対策懇談会において、「子供のころから発症し死に至る可能性がある」として、新たな認定としては約4年振りに、進行性骨化性線維異形成症を国指定の難病として認定することを決めた。
病状診断
数年に渡って骨組織が増殖して関節を固定するため、患者は歩くことや食事、呼吸さえも自力で出来なくなってしまう。この疾患は一般に骨化することで内部組織を押しつぶし、最終的には死に至ることになる。患者は30歳までに身体を動かすことが出来なくなり40歳以上命を長らえさせることは稀である。現在の段階では治療不能の病気と考えられている。
症状
- 短く大きな足の親指が特徴である。
- 生まれつきの外反母趾。
- 進行性骨化性線維異形成症骨組織を構成するに至る最初の症状は10歳前に起こる。
- 腫瘍状の塊が一夜の内に発生することが多い。
- 初期はガンや線維腫と誤診されることもある。病変部の生検はしばしば最初に行われる検査であるが、生検査自体の刺激により病変部位が増大することがある。
症例
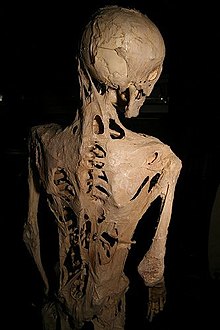
1800年代以降、医学文献には文字どおり「石化」と表現されていたが、現在はこれらの症状の原因をFOPと見なすことが可能である。
著名なFOPの患者としては、1933年12月にフィラデルフィアで生まれたハリー・レイモンド・イーストラックがいる。彼の症状は10歳で進行し、1973年に40歳の誕生日を迎える1週間前、肺炎で死に至った。彼の身体は完全に骨化しており、わずかに唇を動かすことのみ可能であった。彼は死の直前、自分の身体を医学の発展のために提供することを望んだため、骨格はフィジシャンズ・オブ・フィラデルフィア大学内のムッター博物館に保存され、FOPの研究に貴重な資料を提供している。
日本では8歳でFOPと診断された男性が、サンプルとして京都大学iPS細胞研究所に自身の皮膚の提供や集めた募金を寄付するなどしている。2017年(平成29年)8月、男性が19歳になった時に、提供された皮膚から作製されたiPS細胞を使って京都大学の研究グループが「ラパマイシン」が進行性骨化性線維異形成症の進行を遅らす効果があることを発見し、治験を始めると発表した。男性は記者会見で治験にも参加したいと述べている。同年10月5日、京都大学病院は「ラパマイシン」を用いた臨床試験を開始したと発表した。これは、iPS細胞を使って発見した薬を用いた世界初の臨床試験となり、皮膚を提供した男性も臨床試験に参加することとなった。
治療法
FOP患者は体を動かす場合に特に気をつけなければならないことがある。それには高い場所から飛び降りたり、打撲をしないように細心の注意をし、骨組織を増殖させる原因となりうる筋肉注射は避けることなどが含まれる。同様に運動の際に日常的に曲げる角度を超えて関節を曲げてはならない。
現時点では、一度症状が現れてから骨増殖を抑える完全な予防治療は存在しない。しかし、将来的には遺伝子治療の可能性はある。
2017年(平成29年)8月1日、京都大学の池谷真准教授と戸口田淳也教授らの研究グループが進行性骨化性線維異形成症の治療薬として「ラパマイシン」をiPS細胞を使って見つけた。また同日、臨床試験を開始すると発表した。iPS細胞を使った創薬の治験は世界で初めてとなる。
原因
常染色体2番染色体の長腕 (q23-24) にある優性対立遺伝子が原因である。
この対立遺伝子は表現度の差異は存在するが完全浸透を示す。FOP患者のほとんどは子供を遺せないので、症例の殆どは健常な両親のどちらかの配偶子に起きた突然変異が原因となっている。発症頻度は200万人に1人(1世代当たり1.8(SE±1.04)×10-6回の割合で変異)。より症状の軽い類縁疾患として線維性骨異形成があるが、こちらは接合後変異(受精より後に起きる突然変異)が原因となる。
この疾患は、ACVR1遺伝子(別名:2型アクチビン様受容体キナーゼ〈Activin-like receptor kinase 2〉遺伝子、ALK2)の変異に起因する。ACVR1遺伝子は、1型骨形成タンパク質受容体の一種である1型アクチビン受容体(=ACVR1タンパク)をエンコードしている。変異により、ACVR1タンパクの第206残基がヒスチジンからアルギニンに置き換わる。これによって内皮細胞が間葉幹細胞に形質転換し更に骨に化生するという異常が起きやすくなる。
脚注
注釈
資料
- The International FOP Association
- "Fibrodysplasia ossificans progressiva" WebMDHealth
- http://jmg.bmjjournals.com/cgi/content/abstract/19/1/35
- http://www.blackwell-synergy.com/links/doi/10.1034/j.1399-0020.1999.285280512.x/abs
- http://www.timesonline.co.uk/article/0,,11069-2148620,00.html
外部リンク
- 進行性骨化性線維異形成症(FOP)(指定難病272) - 難病情報センター
- FOP(進行性骨化性線維異形成症)の異所性骨化部の起源は? 慶應義塾大学