
| |

| |
| IUPAC命名法による物質名 | |
|---|---|
| |
| 臨床データ | |
| 販売名 | Casodex, others |
| Drugs.com | monograph |
| MedlinePlus | a697047 |
| ライセンス | US FDA:リンク |
| 胎児危険度分類 | |
| 法的規制 |
|
| 投与方法 | By mouth |
| 薬物動態データ | |
| 生物学的利用能 | Well-absorbed; absolute bioavailability unknown |
| 血漿タンパク結合 |
Racemate: 96.1% (R)-Isomer: 99.6% (Mainly to albumin) |
| 代謝 |
Liver (extensively): • Hydroxylation (CYP3A4) • Glucuronidation (UGT1A9) |
| 代謝物質 | • Bicalutamide glucuronide • Hydroxybicalutamide • Hydroxybicalutamide gluc. (All inactive) |
| 作用発現 | Unknown |
| 作用持続時間 | Unknown |
| 識別 | |
| 別名 | ICI-176,334; ZD-176,334 |
| 化学的データ | |
| 化学式 | C18H14F4N2O4S |
| 分子量 | 430.37 g·mol−1 |
| |
| |
| 物理的データ | |
| 融点 | 191 - 193 °C (376 - 379 °F) (experimental) |
| 沸点 | 650 °C (1,202 °F) (predicted) |
| 水への溶解量 | 0.005 mg/mL (20 °C) |
ビカルタミド(Bicalutamide)は、カソデックス(Casodex)など商品名で販売されている主に前立腺癌の治療に用いられる抗アンドロゲン薬である。通常、ゴナドトロピン放出ホルモン(GnRH)アナログまたは睾丸摘出手術と併用して進行性前立腺癌の治療に用いられる。ビカルタミドは、女性の過度の発毛、トランスジェンダーの女性の女性化ホルモン療法の構成要素、男児の思春期早発症の治療、男性の持続勃起症の予防にも用いられる。投与法は経口である。
男性に見られる一般的な副作用には、乳房の肥大、乳房の圧痛、ほてりなどが挙げられる。男性に見られるその他の副作用には、女性化と性機能障害がある。ビカルタミドは女性にはほとんど副作用を引き起こさないようであるが、女性への使用について食品医薬品局(FDA)は推奨していない。日本では女性には禁忌である。妊娠中の人への投与は胎児に害を及ぼす可能性がある。ビカルタミドは、約1%の人に肝酵素の上昇を引き起こす。稀に、肝障害、肺毒性、光線過敏症の症例に関連している。肝臓への悪影響のリスクは少ないが、治療中は肝機能を監視することが勧められている。
ビカルタミドは、非ステロイド系抗アンドロゲン薬(NSAA)に属する医薬品の一つである。ビカルタミドの作用機序は、アンドロゲンホルモンであるテストステロンとジヒドロテストステロン(DHT)の生物学的標的であるアンドロゲン受容体(AR)を遮断することによって機能する。アンドロゲンレベルを下げることはない。この医薬品は男性にエストロゲンのような効果をもたらす可能性がある。ビカルタミドは吸収され易く、食物によって吸収率が左右されることはない。ビカルタミドの消失半減期は約1週間である。血液脳関門を通過すると考えられ、体と脳の両方に影響を与える。
ビカルタミドは1982年に特許を取得し、1995年に医療用として承認された。日本では1999年3月に承認された。WHO必須医薬品モデル・リストに収載されている。日本でも後発医薬品として入手可能である。開発途上国の卸売り価格は1か月分で約7.07 - 144.22米ドルである。米国では、1か月分で10米ドル以上の費用が掛かる。ビカルタミドは先進国を含む80以上の国で販売されている。ビカルタミドは、前立腺癌の治療に最も広く使用されている抗アンドロゲンであり、何百万人もの前立腺癌疾患の男性に処方されている。
効能・効果
日本で承認されている効能・効果は「前立腺癌」であり、併用薬・併用療法に制限はない。除睾術と併用されたり単剤で用いられたりしている。
米国などではビカルタミドは、主に以下の効能・効果で承認されている。
- 男性における転移性前立腺癌(mPC)に対する性腺刺激ホルモン放出ホルモン(GnRH)アナログ製剤または外科的去勢との併用療法
- 男性における局所進行性前立腺癌(LAPC)の単剤療法(米国では未承認)
また、ビカルタミドは以下のような適応症でも採用されている(非承認)。
- 男性におけるGnRHアゴニスト治療開始時のテストステロン・フレアの影響の軽減
- 女性のにきび、脂漏、多毛、頭皮脱毛などのアンドロゲン依存性の皮膚・毛髪疾患、および女性の多嚢胞性卵巣症候群(PCOS)によるテストステロン高値。避妊用ピル併用。
- トランスジェンダー女性に対する女性化ホルモン療法。エストロゲン併用。
- 男児の末梢性思春期早発症、特に家族性男性限定思春期早発症(精巣中毒症)の場合。アナストロゾールなどのアロマターゼ阻害剤併用。
- 男性の持続勃起症
この薬は以下の適応症が示唆されているが、有効性は不明である。
これらの用途の詳細については、英語版「Medical uses of bicalutamide」を参照
禁忌
ビカルタミドは日本では、小児と女性に禁忌とされている。また、米国では妊娠カテゴリーX(妊娠禁忌)、オーストラリアでは妊娠カテゴリーD(2番目に制限されたランク)に分類されている。すなわち、妊娠中の女性には禁忌であり、性的に活発で妊娠する可能性のある女性は、適切な避妊法と組み合わせてのみビカルタミドを服用することが強く推奨される。ビカルタミドが母乳中に排泄されるかどうかは不明だが、多くの薬剤は母乳中に排泄されるため、授乳中は同様にビカルタミドの治療は推奨されない。
重度の肝機能障害を持つ人ではビカルタミドの排泄が遅くなるという証拠があり、従ってこれらの患者ではビカルタミドの循環レベルが上昇する可能性があり、注意が必要とされる。重度の肝障害では、ビカルタミドの活性型(R)エナンチオマーの排泄半減期が約1.75倍(76%増、排泄半減期は健常者で5.9日、障害者で10.4日)に増加する。腎障害では、ビカルタミドの排泄半減期は変化しない。
副作用
重大な副作用は、
である。
5%以上の患者に乳房腫脹が現われる。
詳細は英語版「Side effects of bicalutamide」を参照。
他の抗アンドロゲン薬との比較
男女におけるビカルタミドの副作用プロファイルは、他の抗アンドロゲン薬とは異なり、比較的に良好であると考えられている。GnRHアナログ薬や、ステロイド系抗アンドロゲン薬(SAA)である酢酸シプロテロン(CPA)と比較して、ビカルタミド単剤療法は、火照りや性機能障害の発生率および重症度が遥かに低い。また、GnRHアナログ製剤やCPAとは異なり、ビカルタミド単剤療法は骨密度の低下や骨粗鬆症とは無縁である。逆に、ビカルタミド単剤療法は、GnRHアナログ製剤やCPAと比較して、男性における乳房圧痛、女性化乳房 、女性化の発生率が非常に高いとされている。しかし、ビカルタミドによる女性化は重度であることは稀であり、この副作用による中止率はかなり低いとされている。こうしたビカルタミド単剤、GnRHアナログ、CPAの副作用の違いは、GnRHアナログやCPAがエストロゲンの産生を抑制するのに対し、ビカルタミド単剤はエストロゲンレベルを低下させず、むしろ増加させるという事実に起因する。
ビカルタミドは、CPAと関連している抑うつや疲労などの精神神経系の副作用や、凝固変化、血栓、体液貯留、虚血性心筋症、有害な血清脂質変化などの心血管系の副作用のリスクを共有していない。ビカルタミドは、フルタミドやCPAよりも肝毒性が、ニルタミドよりも間質性肺炎のリスクが遥かに低いまた、フルタミドの下痢、ニルタミドの悪心・嘔吐・視覚障害・アルコール不耐性といった特有のリスクもない。エンザルタミドとは異なり、ビカルタミドは痙攣発作や、不安や不眠といった中枢性の副作用とは無縁である。しかし、ビカルタミドによる肝臓の有害な変化のリスクは低いが、エンザルタミドはビカルタミドと異なり、肝酵素の上昇や肝毒性のリスクが知られていない。ステロイド系抗アンドロゲン薬であるスピロノラクトンとは対照的に、ビカルタミドは抗鉱質コルチコイド作用を有さず、そのため高カリウム血症、頻尿、脱水、低血圧、その他の関連する副作用とは無縁である。女性では、CPAやスピロノラクトンと異なり、ビカルタミドは月経不順や無月経を起こさず、排卵や受胎能力を妨げない。
過量投与
日本で承認されている用量は1日1回80mgである。米国などでは基本的に1日1回50mgである。ヒトにおけるビカルタミドの単回経口投与で、過量投与の症状が現れたり、生命を脅かすと考えられる用量は確立されていない。臨床試験では600mg/日までの投与量で良好な忍容性が得られており、ビカルタミドの吸収が飽和しているため、活性 (R)-エナンチオマーの血中循環濃度は300mg/日の投与量を超えてもそれ以上上昇しないことが注目されている。ビカルタミドや他の第一世代のNSAA(フルタミド、ニルタミドなど)では、過量投与による生命への影響はないと考えられている。79歳の男性がニルタミドを大量に過量摂取した際(13g、通常の最大臨床投与量である300mg/日の43倍)、臨床的な徴候、症状、毒性は見られず、問題は起こらなかった。ビカルタミドまたはNSAAの過量投与に対する特異的な解毒剤はなく、症状がある場合はその症状に基づいて治療を行う必要がある。
相互作用
ビカルタミドは、ほとんどがCYP3A4によって代謝される。そのため、CYP3A4の阻害薬や誘導薬によって体内の濃度が変化する可能性がある。しかし、ビカルタミドはCYP3A4で代謝されるにもかかわらず、150mg/日以下の用量のビカルタミドとシトクロムP450酵素活性を阻害または誘導する薬剤を併用しても、臨床的に重大な薬物相互作用の証拠は観察されなかった。
ビカルタミドは比較的高濃度で循環し、かつタンパク質との結合性が高いため、ワルファリン、フェニトイン、テオフィリン、アスピリンなどのタンパク質との結合性が高い薬剤が血漿中の結合タンパク質から追い出される可能性がある。その結果、これらの薬剤の遊離濃度が上昇し、効果や副作用が増強され、投与量の調整が必要になる可能性がある。特にビカルタミドは、ワルファリンなどのクマリン系抗凝固剤を血漿中の結合タンパク質(すなわちアルブミン)から追い出し、抗凝固作用が増強される可能性があることがin vitro で確認されており、このため、ビカルタミドをこれらの薬剤と併用する場合には、プロトロンビン時間を注意深く観察し、必要に応じて投与量を調整することが推奨されている。しかし、それにもかかわらず、約3,000人の患者を対象とした臨床試験において、ビカルタミドと他の薬剤との相互作用を示す決定的な証拠は発見されていない。
薬理学的特性
薬理作用
抗アンドロゲン活性
ビカルタミドは、アンドロゲン性ホルモンであるテストステロンとDHTの主要な生物学的標的であるアンドロゲン受容体(AR)に対して高選択的競合的完全遮断薬(IC50 = 159 - 243 nM)として作用し、従って抗アンドロゲン作用を有する。ビカルタミドの活性は、(R)-異性体にある。ARに対する選択性のため、ビカルタミドは他のステロイドホルモン受容体と重要な相互作用を示さず、臨床的にはオフターゲットのホルモン活性(例:黄体ホルモン、エストロゲン、糖質コルチコイド、抗鉱質コルチコイド)はない。しかし、ビカルタミドは、アンタゴニストであるプロゲステロン受容体(PR)に弱い親和性を示すことが報告されており、そのため、何らかの抗黄体ホルモン作用を有する可能性があるとされている。ビカルタミドは、5α-リダクターゼを阻害せず、アンドロゲンのステロイド生成に関与する他の酵素(CYP17A1など)を阻害することも知られている。ビカルタミドはエストロゲン受容体(ER)には結合しないが、男性に単剤で使用した場合、AR阻害により二次的にエストロゲン量を増加させる可能性があり、男性において間接的なエストロゲン作用を示す可能性がある。ビカルタミドは、体内のアンドロゲン産生を抑制も阻害もしない(すなわち、抗ゴナドトロピンやアンドロゲンのステロイド産生阻害薬として作用したり、アンドロゲンレベルを低下させたりしない)ため、もっぱらARに拮抗することで抗アンドロゲン作用を発揮する。古典的な核内ARに加えて、ビカルタミドは膜型アンドロゲン受容体(mAR)でも評価され、ZIP9の強力なアンタゴニスト(IC50 = 66.3 nM)として作用することが判明したが、GPRC6Aとは相互作用しないと思われた。
ビカルタミドのARに対する親和性は、バイオアッセイにおいてテストステロンの2.5 - 10倍のARアゴニストであり、前立腺における主な内因性リガンドであるDHTの親和性の約30 - 100分の1と比較的低い値を示す。しかし、ビカルタミドの一般的な臨床投与量では、テストステロンやDHTの数千倍の血中濃度となり、これらが受容体に結合して活性化するのを強力に阻止することができる。これは、外科的または内科的な去勢を行った場合に特に顕著で、循環血液中のテストステロン濃度は約95%減少し、前立腺中のDHT濃度は約50 - 60%減少する。女性の場合、テストステロン濃度は男性に比べて大幅に低い(20 - 40倍)ため、より少量のビカルタミド(例えば、多毛症に対する臨床試験では25mg/日)が必要となる。
下垂体および視床下部のARをビカルタミドで遮断すると、男性の場合、アンドロゲンの視床下部-下垂体-性腺軸(HPG軸)への負のフィードバックが阻止され、その結果、下垂体の黄体形成ホルモン(LH)の分泌が脱抑制される。その結果、循環LHレベルが上昇し、性腺でのテストステロンの産生が活性化され、ひいてはエストラジオールの産生も活性化される。150mg/日のビカルタミド単剤投与により、テストステロンは1.5 - 2倍(59 - 97%)、エストラジオールは1.5 - 2.5倍(65 - 146%)に増加することが確認されている。テストステロンおよびエストラジオールに加えて、DHT、性ホルモン結合グロブリンおよびプロラクチンの濃度にも小さな増加が認められた。エストラジオール濃度は閉経前女性の正常範囲下限と近い値を示しており、一方テストステロン濃度は一般的に男性の正常範囲の上限に位置している。テストステロン濃度は、エストラジオール濃度の上昇によるHPG軸への負のフィードバックにより、通常、男性の正常範囲を超えることはない。ビカルタミドがHPG軸に影響を与え、ホルモン濃度を上昇させるのは男性のみで、女性には影響しない。これは、女性のアンドロゲン濃度が非常に低く、基礎的なHPG軸の抑制が行われないためである。前立腺癌などのアンドロゲン依存性疾患の治療に有効であることからもわかるように、ビカルタミドの抗アンドロゲン作用は、結果として生じるテストステロンの増加に因る影響を大幅に上回る。しかし、エストラジオールの増加はビカルタミドによって抑制されないため、女性化乳房や雌性化の原因となる。ビカルタミドの単剤投与は、男性のゴナドトロピンおよび性ホルモンレベルを上昇させるが、ビカルタミドとGnRHアナログなどの抗ゴナドトロピン薬、エストロゲン、プロゲストーゲンを併用すると、HPG軸への負のフィードバックが維持されるため、この現象は起こらない。
ビカルタミドを含むNSAA単剤療法は、外科的・内科的な去勢を伴うアンドロゲン遮断療法の方法とは、多くの忍容性の違いがある。例えば、GnRHアナログ製剤では、ほてり、抑うつ、疲労、性機能障害などの発現率がNSAA単剤療法に比べて非常に高いことが分かっている。これは、GnRHアナログ製剤が、アンドロゲンに加えてエストロゲンの産生も抑制し、エストロゲン欠乏症を引き起こすためと考えられている。一方、NSAA単独療法では、エストロゲンレベルは減少せず、むしろ増加するため、エストロゲンが過剰に分泌され、アンドロゲンの欠乏を補い、気分、エネルギー、性機能の維持が可能となる。また、3α-アンドロスタンジオールや3β-アンドロスタンジオールのようなテストステロンから生成される神経ステロイドは、ERβアゴニストであり、前者は強力なGABAA受容体陽性アロステリック調節因子であるため、関与している可能性がある。性機能障害の具体的なケースでは、アンドロゲン産生抑制薬を併用しなければ、脳内のビカルタミドによるARの遮断が不完全であり、性的機能に顕著な影響を与えるには不十分であることが、この違いの追加的な可能性として考えられる。
通常の環境下では、ビカルタミドはARを活性化する能力を持たない。しかし、前立腺癌では、ARの変異や過剰発現が前立腺細胞に蓄積し、ビカルタミドがARのアンタゴニストからアゴニストへと変わることがある。この結果、ビカルタミドが前立腺癌の成長を逆説的に促進することになり、抗アンドロゲン剤を中止すると前立腺癌の成長速度が逆説的に遅くなる抗アンドロゲン薬離脱症候群という現象の原因となっている。
ビカルタミドの単剤投与は、精巣の精子形成、精巣の微細構造、および男性の生殖能力の一部にほとんど影響を及ぼさないようである。これは、精巣(男性のテストステロンの約95%が産生される場所)のテストステロン濃度が非常に高く(血中濃度の200倍)、精子形成を維持するために実際に必要な精巣のテストステロン濃度は正常値の極一部(10%未満)に過ぎないためと考えられる。そのため、ビカルタミドは、この精巣でテストステロンと競合し、アンドロゲンのシグナル伝達や機能を阻害することはできないと考えられる。しかし、ビカルタミドは精巣の精子形成には影響を与えないとおぼしき一方で、精巣以外の精巣上体や精管でのAR依存性の精子の成熟や輸送を阻害する可能性があり、その結果、男性の生殖能力に障害を与える可能性がある。さらに、エストロゲン、プロゲストーゲン、GnRHアナログなどの他の薬剤とビカルタミドを併用すると、それぞれの薬剤が男性の生殖能力に悪影響を及ぼすため、精子形成が損なわれる可能性がある。これらの薬剤は性腺のアンドロゲン産生を強く抑制することができ、精巣の精子形成を著しく損なったり、消失させたりする可能性がある。また、エストロゲンは、十分に高い濃度で精巣に直接的かつ長期的な細胞毒性を及ぼす可能性がある。
その他の活性
ビカルタミドは、前臨床研究において、CYP3A4、CYP2C9、CYP2C19、CYP2D6を含む特定のシトクロムP450酵素の阻害薬または誘導薬として作用することが確認されているが、最大150mg/日の投与を受けたヒトにおいては、その証拠は見いだされなかった。また、in vitro では、CYP27A1(コレステロール27水酸化酵素)の強力な阻害剤として、また、CYP46A1(コレステロール24水酸化酵素)の阻害剤として同定されているが、in vivo またはヒトでの評価・確認はまだ行われておらず、臨床的意義は不明である。ビカルタミドは、P-糖タンパク質(ABCB1)阻害剤であることが判明している。他の第一世代のNSAAやエンザルタミドと同様に、in vitro でGABAA受容体を介した電気信号に対して弱い非競合的阻害剤として作用することが確認されている(IC50=5.2μM)。しかし、エンザルタミドとは異なり、ビカルタミドには痙攣などの中枢性の副作用は認められておらず、この知見の臨床的意義は不明である。
薬物動態
ヒトにおける絶対的なバイオアベイラビリティーは不明であるが、ビカルタミドは広範かつ良好に吸収されることが知られている。その吸収は食事の影響を受けない。ビカルタミドの吸収は、150mg/日までは直線的で、それ以上の用量では飽和状態となり、300mg/日以上の用量ではビカルタミド濃度は定常状態でそれ以上上昇しない。(R)-ビカルタミドの吸収は遅く、投与後31 - 39時間でピークに達するのに対し、(S)-ビカルタミドは吸収が非常に速い。投与量に関係なく、4 - 12週間の投与で定常濃度に達し、(R)-ビカルタミドの濃度は10 - 20倍に増加する。定常濃度に達するまでの期間が長いのは、ビカルタミドの排泄半減期が非常に長いことに起因する。
ビカルタミドの組織分布は十分に明らかにされていない。性交時にパートナーの女性に移行する可能性のある精液中のビカルタミドの量は少なく、重要ではないと考えられている。ラットやイヌを用いた動物実験では、ビカルタミドは血液脳関門を通過しないため、脳には入らないと考えられていたので、当初は末梢選択的な抗アンドロゲン剤だと考えられていた。しかし、その後の臨床試験で、ヒトではそうではないことが判明し、種の違いが示された。ビカルタミドはヒトの脳内に入り、それに伴い中枢性抗アンドロゲン作用に合致した効果と副作用を発揮する。ビカルタミドは血漿タンパク質との結合率が高く(ラセミ体で96.1%、(R)-ビカルタミドで99.6%)、主にアルブミンと結合し、性ホルモン結合グロブリンや糖質ステロイド結合グロブリンとの結合は極僅かである。
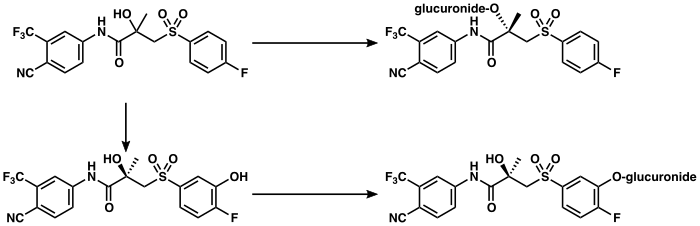
ビカルタミドは肝臓で代謝される。(R)-ビカルタミドは、CYP3A4による水酸化を介してゆっくりと、ほぼ独占的に代謝され、(R)-ヒドロキシビカルタミドになる。この代謝物は、UGT1A9によってグルクロン酸抱合体に変換される。(R)-ビカルタミドとは対照的に、(S)-ビカルタミドは速やかに、主にグルクロン酸抱合で代謝される(水酸化は行われない)。ビカルタミドの代謝物はいずれも活性がないことが知られており、血漿中では代謝物のレベルは低く、未変化のビカルタミドが優勢である。ビカルタミドは立体選択的に代謝されるため、(R)-ビカルタミドは (S)-ビカルタミドに比べて最終的な半減期が非常に長く、その濃度は単回投与で約10 - 20倍、定常状態では約100倍になる。(R)-ビカルタミドの排泄半減期は、単回投与で5.8日、反復投与で7 - 10日と比較的長い。
ビカルタミドは糞中(43%)と尿中(34%)に概ね同程度の割合で排泄され、代謝物も尿中と胆汁中にほぼ均等に排泄される。ビカルタミドはかなりの部分が未代謝の状態で排泄され、未変化体と代謝物のグルクロン酸抱合体も排泄される。ビカルタミドとその代謝物のグルクロニド抱合体は、非抱合体のビカルタミドとは異なり、速やかに循環系から排除される。
ビカルタミドの薬物動態は、食物の摂取、年齢や体重、腎機能障害、軽度から中等度の肝機能障害の影響を受けない。しかし、日本人では白人に比べてビカルタミドの定常濃度が高い。
化学的特徴
ビカルタミドは,エナンチオマーである (R)-ビカルタミド(右旋性)と (S)-ビカルタミド(左旋性)からなるラセミ混合物である。
ビカルタミドはフルタミドから派生した合成の非ステロイド性化合物である。ビシクロ化合物(2つの環を持つ)であり、アニリド(N-フェニルアミド)またはアニリン、ジアリルプロピオンアミド、トルイジン類に分類され、さまざまに言及されている。
臨床試験
ビカルタミドは1987年に最初の第I相臨床試験が行われ、前立腺癌における最初の第II相臨床試験の結果が1990年に発表された。ICIの製薬部門は1993年にゼネカという独立した会社に分割され、1995年4月と5月にゼネカ(現在のアストラゼネカ、1999年にアストラABと合併後)が前立腺癌の治療のためにビカルタミドの米国での承認前マーケティングを開始した。1995年5月に英国で発売され、続いて1995年10月4日に米国FDAより、GnRHアナログと併用して50mg/日の用量で前立腺癌の治療薬として承認された。
GnRHアナログとの併用療法が導入された後、ビカルタミドは、前立腺癌の治療のために150mg/日の用量で単剤療法として開発され、1990年代後半から2000年代前半にかけて、ヨーロッパ、カナダおよびその他の多くの国でこの適応症で承認された。ビカルタミドのこの用途は、2002年に米国でもFDAによって審査されたが、最終的には承認されなかった。日本では、前立腺癌の治療薬として、ビカルタミドを80mg/日の用量で単独またはGnRHアナログと併用することが許可されている。日本で使用されているビカルタミドの独自の80mgの用量は、日本人男性におけるビカルタミドの薬物動態の違いが観察されたことに基づいて、日本での開発のために選択された。
早期前立腺癌の試験において、非転移性前立腺癌に対するビカルタミド単剤療法が否定的な結果となったことを受け、英国(2003年10月または11月)、その他の欧州諸国およびカナダ(2003年8月)を含む多くの国で、非転移性前立腺癌治療に特化したビカルタミドの承認が取り消された。また、米国およびカナダでは、本適応症に対する150mg/日のビカルタミドの使用を明確に推奨していた。一方で、ビカルタミドは局所進行前立腺癌および転移性前立腺癌の治療に有効であり、引き続き承認され、使用されている。
外部リンク
- “Bicalutamide”. Drug Information Portal. U.S. National Library of Medicine. 2021年5月27日閲覧。