






嚢胞性線維症膜コンダクタンス制御因子(のうほうせいせんいしょうまくコンダクタンスせいぎょいんし、英: cystic fibrosis transmembrane conductance regulator、略称: CFTR)は、脊椎動物に存在する膜タンパク質かつ塩素チャンネルであり、CFTR遺伝子にコードされる。
CFTR遺伝子はABC輸送体型のイオンチャネルをコードし、上皮細胞の細胞膜を越えて塩化物イオンを透過させる。塩化物イオンチャネル機能に影響を与えるCFTR遺伝子の変異は肺、膵臓などの器官の上皮を覆う液体の輸送の調節異常をもたらし、嚢胞性線維症を引き起こす。合併症としては、高頻度の呼吸器感染症を伴う肺粘液の粘性の増加や、膵臓の機能不全による栄養不良や糖尿病などが挙げられる。これらの症状は、慢性的な障害や寿命の短縮につながる。男性では、発生中の精管(精索)や精巣上体で管内分泌の異常による閉塞と破壊が進行し、先天性精管欠損症や男性不妊の原因となるようである。
遺伝子

ヒトのCFTRをコードする遺伝子は7番染色体長腕q31.2、116,907,253番から117,095,955番の塩基対に位置する。CFTRのオルソログは顎口上綱に存在する。
動物のCFTR遺伝子は核DNAの系統遺伝学的マーカーとして利用される。この遺伝子の長大なゲノム配列は哺乳類の主要なグループの系統学的関係の探索に用いられ、有胎盤類の異節上目、アフリカ獣上目、ローラシア獣上目、真主齧上目という4つの大きなクレードへの分類を確証した。
変異
嚢胞性線維症(CF)を引き起こす変異は1000種類近く記載されている。最も一般的な変異はΔF508であり、3つのヌクレオチドの欠失(Δ)によって、タンパク質の508番目のアミノ酸であるフェニルアラニン(F)が失われる。その結果、タンパク質は正常なフォールディングが行われず、より早く分解される。他の大部分の変異の頻度は低いが、変異の分布と頻度は集団によって異なり、遺伝学的スクリーニングやカウンセリングに影響を与える。
病気の原因となる変異の数が多いため、CFのすべての患者を対象とした治療薬の創出は困難である。広範囲に作用する薬剤候補のスクリーニングを行うためには、理想的にはすべての変異体に対応する細胞株と細胞ベースのアッセイのライブラリが必要となる。各変異体に対応するクローン細胞の検出・単離には、蛍光オリゴヌクレオチドシグナルプローブなどの細胞工学的手法を用いることができる。
変異にはCFTR遺伝子の塩基置換、重複、欠失や短縮がある。その結果、タンパク質は全く機能しないものや効率的に機能しないものとなったり、より迅速に分解されたり、十分量が存在しないものとなったりする。
CFTR遺伝子の変異は、ヘテロ接合型の個体に選択的優位性を与えている可能性がある。変異型CFTRタンパク質を発現する細胞は、腸チフスの原因菌であるサルモネラの侵入に抵抗性を示し、変異型CFTRを1コピーだけ持つマウスはコレラ毒素による下痢に抵抗性を示す。
コーカソイドで最も一般的な変異には次のようなものがある。
- ΔF508
- G542X
- G551D
- N1303K
- W1282X
ΔF508
ΔF508(CFTRΔF508、F508del-CFTR、rs113993960)は、7番染色体上のCFTR遺伝子の507番から508番のアミノ酸をコードする領域で3つのヌクレオチドが欠失した変異であり、CFTRタンパク質からは508番のフェニルアラニンが失われる。ΔF508変異によって産生されるフェニルアラニン残基を欠いた異常なCFTRタンパク質は適切なフォールディングが行われない。このタンパク質はさらなるプロセシングのための小胞体からの移行が起こらない。この変異を2コピー持つ(両親から1コピーずつ受け継ぐ)ことが嚢胞性線維症(CF)の最も一般的な原因であり、世界中で観察されている変異の約2/3を占める。
影響
CFTRタンパク質は主に膵臓、腸、呼吸上皮、外分泌腺で発現している。適切なフォールディンが行われた場合には、CFTRは細胞膜へ移行して膜タンパク質となり、塩化物イオンを細胞外へ放出するチャネルの開口を担うとともに、他のチャネルタンパク質によるナトリウムイオンの取り込みも同時に阻害する。これらの機能は、水を細胞外へ運ぶ浸透圧を形成するイオン勾配の維持に寄与する。ΔF508変異はCFTRの誤ったフォールディングを引き起こし、タンパク質は最終的には小胞体内で分解される。この変異を2コピー抱える個体では、CFTRタンパク質は細胞膜に全く存在しなくなり、重要なイオン輸送機能が行われなくなる。
ホモ接合型ΔF508変異では、CFTRタンパク質は細胞膜の正しい位置に存在しなくなる。その結果、細胞内の水分保持量は増加し、それに伴って細胞外は脱水状態となり、体のさまざまな部分で影響の連鎖が生じる。そうした影響としては、器官の上皮の粘膜の肥厚、粘液の粘性の増加と粘膜線毛系の自由な運動の阻害による気道の閉塞、胎児の発生過程での粘液の粘性の増加による先天性精管欠損症、粘液による膵管の閉塞による膵臓の機能不全、細菌が繁殖する栄養豊富で濃厚な粘液の蓄積による呼吸器感染症のリスクの増加などがある。これらは遺伝子疾患である嚢胞性線維症の症状であるが、ΔF508がこの疾患の原因となる唯一の変異であるわけではない。
ヘテロ接合型ΔF508変異(ΔF508変異を1コピーだけ持つ)では、CFTRの機能不全または不在のために細胞膜を挟んだ安定なイオン勾配を維持することができず、一方でこのことは下痢の際の水分喪失の低下をもたらす。一般的には、Cl−とNa+の双方が細胞内に蓄積するため細胞外が低浸透圧となり、水は浸透によって細胞内へ拡散する。いくつかの研究では、ヘテロ接合型変異の保因者はさまざまな症状のリスクが高いことが示されている。例えば、嚢胞性線維症の原因となる変異のヘテロ接合型としての保有は気道の反応性の増加と関連していることが示されており、肺機能の低下のリスクがある可能性がある。また、喘鳴を伴うヘテロ接合型変異は、肺機能の低下や慢性閉塞性肺疾患の発症と進行のリスクが高いことが示されている。嚢胞性線維症の原因となる遺伝子が1つでもあれば、感染がない場合でも肺に軽度の異常が生じる可能性がある。
機構
CFTR遺伝子は7番染色体長腕q31.2に位置し、1480アミノ酸からなる配列をコードする。通常、507番目のアミノ酸に対応するコドンのDNA配列はATC(相補鎖の3'-TAG-5'と対合する)であり、mRNA上ではAUCとなってイソロイシンをコードする。そして508番目はフェニルアラニンをコードするTTT(相補鎖は3'-AAA-5')で、mRNA上ではUUUとなってフェニルアラニンをコードする。ΔF508変異では、507番目のコドンの3番目C-G塩基対と508番目のコドンの最初の2つのT-A塩基対が欠失し、ATTのDNA配列が残される。ATTはmRNA上ではAUUとなり、AUUはイソロイシンをコードするため、507番目のアミノ酸は変化せず、508番目のアミノ酸であるフェニルアラニンが欠失した形となる。
有病率
コーカソイドでは、約30人に1人が少なくとも1コピーのΔF508変異を保有している。CFTR遺伝子の双方のコピーにこの変異が存在すると、常染色体劣性遺伝疾患である嚢胞性線維症(CF)が引き起こされる。この変異は52,000年以上前に北ヨーロッパで生じたと推定されている。若いallele age(変異が最初に出現してからの期間)は過去の自然選択の結果である可能性がある。このような有害な変異が自然選択の過程で維持されてきた理由として、この変異が1コピーだけ存在する場合、コレラに感染した際の水分損失が軽減されるというプラスの効果をもたらされる可能性があるという仮説が立てられているものの、病原性のあるコレラ菌Vibrio choleraeがヨーロッパにもたらされたのは18世紀後半である。他の仮説としては、CFTRはサルモネラが腸管上皮細胞へ進入する際の受容体として働くことから、ΔF508のヘテロ接合型保因者は腸チフスに対してより高い耐性があるとする説がある。
ΔF508のヘテロ接合型変異は喘息の患者に多くみられ、非保因者に比べて肺機能が低い可能性がある。CFの原因となる変異のヘテロ接合型保因者は、一般集団と比較して慢性副鼻腔炎の有病率が高い。ヨーロッパにおけるCFの症例の約50%はΔF508のホモ接合型変異によるものであり(この値は地域によって大きく異なる)、ΔF508のアレル頻度は約70%である。残りの症例は、R117H、1717-1G>A、2789+56G>Aなど、他の1500種類以上の変異が原因となっている。これらの変異が互いに組み合わされた場合や、あるいはΔF508が1コピーであっても、CFの症状が引き起こされる可能性がある。特定の症状と特定の変異との関連づけは行われているものの、遺伝子型とCFの重症度との間に強い相関関係はみられない。
構造

CFTR遺伝子の長さは約189 kbであり、27個のエクソンと26個のイントロンからなる。CFTRタンパク質は1480アミノ酸からなる糖タンパク質である。タンパク質は5つのドメインから構成される。膜貫通ドメイン(TMD)は2つ存在し(TMD1、2)、それぞれ6本のαヘリックスから構成される。これらは細胞質に位置するヌクレオチド結合ドメイン(NBD1、2)によって連結されている。NBD1は調節ドメイン(Rドメイン)によってTMD2と連結されている。RドメインはCFTR特有の構造であり、他のABC輸送体には存在しない。イオンチャネルは、RドメインがPKAによってリン酸化され、NBDにATPが結合しているときにだけ開く。タンパク質のC末端はPDZ相互作用ドメインによって細胞骨格に固定されている。変異を有するNBD1の構造(PDB: 1XMI)では、ホモ五量体に組み立てられることが示されている。
位置と機能
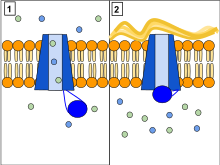
CFTRの機能はリン酸化とATPに依存したアニオンチャネルであり、特定のアニオン(Cl−など)のコンダクタンスを増加させ、電気化学的勾配に従って移動させる。ATPによって駆動されるCTFRのコンフォメーション変化は、アニオンが電気化学的勾配に従って膜を越えて移動する際のゲートを開いたり閉じたりする。これは他のABC輸送体とは対照的であり、これらではATPによって駆動されるコンフォメーション変化によって基質の電気化学的勾配に逆らった、膜を越えた移動が行われる。本質的には、CFTRは開いたコンフォメーション状態で「漏れる」、「壊れた」ABC輸送体として進化したイオンチャネルである。
CFTRには2つのTMDが存在し、それぞれNBDと連結されている。CFTRにはRドメインと呼ばれる他のドメインも存在する。ABC輸送体スーパーファミリーの他のメンバーは、原核生物で栄養素の取り込みに関与していたり、真核生物でさまざまな基質の搬出に関与していたりする。ABC輸送体は、ATPの加水分解による自由エネルギーを、基質の電気化学的勾配に逆らった、細胞膜を挟んだ移動に変換するために進化したものである。ABC輸送体には2つの主要なコンフォメーションが存在し、1つは積み荷結合部位が細胞質側を向いた内向きの状態(ATP非結合状態)、もう1つは外向きの状態(ATP結合状態)である。各NBDへのATPの結合はNBDの二量体化を引き起こし、膜貫通ヘリックスの再配置をもたらす。これによって積み荷結合部位のアクセス性が内向きから外向きへ変化する。ATPの結合とその後の加水分解によって内向きと外向きの積み荷結合部位が交互に露出することで、電気化学的勾配に逆らった一方向への積み荷の輸送が保証される。CFTRでは、内向きコンフォメーションと外向きコンフォメーションを繰り返すことでチャネルの開口と閉口が行われる。特に、NBDの二量体化(ATP結合によって有利となる)は外向きコンフォメーションへの移行と共役しており、アニオンのための開いた膜貫通経路が形成される。その後の加水分解によってNBD二量体は不安定化され、内向きコンフォメーションが有利な状態となり、アニオン透過性経路は閉じられる。
CFTRは、肺、肝臓、膵臓、消化管、女性器、男性器など多くの器官の上皮細胞に存在する。
肺の気道では、CFTRは肺塩類細胞(pulmonary ionocyte)と呼ばれる少数の特殊な細胞で最も高度に発現している。皮膚では、CFTRは皮脂腺とエクリン腺で強く発現している。エクリン腺では、CFTRは汗管を構成する上皮細胞の頂端膜に位置している。
通常、CFTRは塩化物イオンやチオシアン酸イオン(負電荷を持つもの)の上皮細胞から気道液や粘液への移動を可能にする。正に帯電したナトリウムイオンも受動的に輸送されて粘液中の総電解質濃度は増加し、浸透圧によって水が細胞外に移動する。
気管支や卵管に並ぶ、運動性の繊毛を持つ上皮細胞では、CFTRは頂端膜に位置しているが繊毛には位置していない。一方、上皮性ナトリウムチャネル(ENaC)は繊毛全長にわたって位置している。
汗腺でのCFTRの欠陥は塩化ナトリウムとチオシアン酸ナトリウムの再吸収の低下を引き起こし、汗の塩分が高くなる。このことは嚢胞性線維症の臨床的に重要な検査である汗試験の基礎となっており、遺伝子スクリーニングでの診断に利用されることも多い。
相互作用
CFTRは次に挙げる因子と相互作用することが示されている。
関連する疾患
- 先天性精管欠損症: 男性で精管が先天的に両側性で欠損する。ほとんどの場合、CFTR遺伝子の一方のコピーに軽度の変異(遺伝子機能の部分的な変化)、もう一方のコピーに嚢胞性線維症を引き起こす変異が生じている。
- 嚢胞性線維症: これまでにCFTR遺伝子に1800種類以上の変異が見つかっているが、これらの大部分は嚢胞性線維症とは関係していない。これらの変異の大部分ではアミノ酸の置換またはCFTR遺伝子のDNAの小さな欠失が生じている。最も一般的な変異はΔF508と呼ばれるもので、CFTRタンパク質の508番目のアミノ酸であるフェニルアラニン(F)が欠失(Δ)したものである。この変異タンパク質は合成直後に分解されるため、細胞膜へ到達することはない。疾患の原因となる変異はすべてチャネルの正常な機能を阻害し、塩分や水分の細胞内外への移動が妨げられる。この閉塞の結果、肺や膵臓などの器官の通路に並ぶ細胞からは異常に濃厚で粘り気のある粘液が産生される。この粘液が気道や腺を塞ぎ、嚢胞性線維症の特徴的な症状を引き起こす。また、繊毛によって除去されるのは希薄な粘液だけであり、濃厚な粘液は除去されないため、細菌が捕捉されて慢性的な感染症の原因となる。
- コレラ: コレラ毒素によって引き起こされるADPリボシル化はcAMPの産生の増加を引き起こし、CFTRチャネルを開いてCl−の過剰分泌をもたらす。Cl−に続いてNa+と水も小腸へ分泌されるため、脱水と電解質の喪失が引き起こされる。
薬剤標的
CFTRは、関連する疾患の治療のための薬剤標的となっている。イバカフトル(商標名: Kalydeco、開発名: VX-770)は、CFTRに特定の変異を有する嚢胞性線維症患者を対象として2012年にFDAの承認を受けた薬剤である。イバカフトルはバーテックス・ファーマシューティカルズが嚢胞性線維症財団と共同で開発した薬剤で、病気の症状ではなく根本的な原因を治療する初めての薬剤である。2012年で最も重要な新薬、"a wonder drug"(驚異の薬)とも呼ばれたが、年間30万米ドル以上の費用がかかる最も高価な薬剤の1つであり、バーテックス社には高額な費用に対する批判もある。
関連文献
- “A clinical perspective of cystic fibrosis and new genetic findings: relationship of CFTR mutations to genotype-phenotype manifestations”. American Journal of Medical Genetics. Part A 116A (3): 262–7. (January 2003). doi:10.1002/ajmg.a.10886. PMID 12503104.
- “The cystic fibrosis transmembrane conductance regulator: an intriguing protein with pleiotropic functions”. Journal of Cystic Fibrosis 1 (1): 13–29. (March 2002). doi:10.1016/S1569-1993(01)00003-0. PMID 15463806.
- “Mutations and sequence variations detected in the cystic fibrosis transmembrane conductance regulator (CFTR) gene: a report from the Cystic Fibrosis Genetic Analysis Consortium”. Human Mutation 1 (3): 197–203. (1992). doi:10.1002/humu.1380010304. PMID 1284534.
- “Cystic fibrosis transmembrane conductance regulator and the etiology and pathogenesis of cystic fibrosis”. FASEB Journal 6 (10): 2775–82. (July 1992). doi:10.1096/fasebj.6.10.1378801. PMID 1378801.
- “Molecular biology of cystic fibrosis”. Molecular Genetic Medicine 3: 33–68. (1993). doi:10.1016/b978-0-12-462003-2.50006-7. ISBN 9780124620032. PMID 7693108.
- “The molecular basis for disease variability in cystic fibrosis”. European Journal of Human Genetics 4 (2): 65–73. (1996). doi:10.1159/000472174. PMID 8744024.
- “CFTR: domains, structure, and function”. Journal of Bioenergetics and Biomembranes 29 (5): 443–51. (October 1997). doi:10.1023/A:1022430906284. PMID 9511929.
- “Differential function of the two nucleotide binding domains on cystic fibrosis transmembrane conductance regulator”. Biochimica et Biophysica Acta 1461 (2): 263–74. (December 1999). doi:10.1016/S0005-2736(99)00162-5. PMID 10581360.
- “Unique presentations and chronic complications in adult cystic fibrosis: do they teach us anything about CFTR?”. Respiratory Research 1 (3): 133–5. (2000). doi:10.1186/rr23. PMC 59552. PMID 11667976.
- “Cystic fibrosis and CFTR”. Pflugers Archiv 443 Suppl 1: S3-7. (2001). doi:10.1007/s004240100635. PMID 11845294.
- “cAMP signaling cascades and CFTR: is there more to learn?”. Pflugers Archiv 443 Suppl 1: S85-91. (2001). doi:10.1007/s004240100651. PMID 11845310.
- “Regulation of the CFTR channel by phosphorylation”. Pflugers Archiv 443 Suppl 1: S92-6. (2001). doi:10.1007/s004240100652. PMID 11845311.
- “Idiopathic pancreatitis related to CFTR: complex inheritance and identification of a modifier gene”. Journal of Investigative Medicine 50 (5): 247S-255S. (September 2002). doi:10.1136/jim-50-suppl5-01. PMID 12227654.
- “[Cystic fibrosis transmembrane conductance regulator (CFTR) gene: mutations and clinical phenotypes]”. Ugeskrift for Laeger 165 (9): 912–6. (February 2003). PMID 12661515.
- “Two novel null mutations in a Taiwanese cystic fibrosis patient and a survey of East Asian CFTR mutations”. American Journal of Medical Genetics. Part A 120A (2): 296–8. (July 2003). doi:10.1002/ajmg.a.20039. PMID 12833420.
- “CFTR mutations and polymorphisms in male infertility”. International Journal of Andrology 27 (5): 251–6. (October 2004). doi:10.1111/j.1365-2605.2004.00485.x. PMID 15379964.
- “The impact of cystic fibrosis and PSTI/SPINK1 gene mutations on susceptibility to chronic pancreatitis”. Clinics in Laboratory Medicine 25 (1): 79–100. (March 2005). doi:10.1016/j.cll.2004.12.007. PMID 15749233.
- “Establishing a diagnosis of cystic fibrosis”. Chronic Respiratory Disease 1 (4): 205–10. (2004). doi:10.1191/1479972304cd044rs. PMID 16281647.
- “Genetic issues in pediatric pancreatitis”. Current Gastroenterology Reports 8 (3): 248–53. (June 2006). doi:10.1007/s11894-006-0083-8. PMID 16764792.
- “Relationships between cystic fibrosis transmembrane conductance regulator, extracellular nucleotides and cystic fibrosis”. Pharmacology & Therapeutics 112 (3): 719–32. (December 2006). doi:10.1016/j.pharmthera.2006.05.010. PMID 16828872.
- “Patterns of GI disease in adulthood associated with mutations in the CFTR gene”. Gut 56 (8): 1153–63. (August 2007). doi:10.1136/gut.2004.062786. PMC 1955522. PMID 17446304.
外部リンク
- GeneReviews/NCBI/NIH/UW entry on CFTR-Related Disorders - Cystic Fibrosis (CF, Mucoviscidosis) and Congenital Absence of the Vas Deferens (CAVD)
- The Cystic Fibrosis Transmembrane Conductance Regulator Protein
- The Human Gene Mutation Database - CFTR Records
- Cystic Fibrosis Mutation Database
- Oak Ridge National Laboratory CFTR Information
- CFTR at OMIM (National Center for Biotechnology Information)
- Overview of all the structural information available in the PDB for UniProt: P13569 (Human Cystic fibrosis transmembrane conductance regulator) at the PDBe-KB.
- Overview of all the structural information available in the PDB for UniProt: P26361 (Mouse Cystic fibrosis transmembrane conductance regulator) at the PDBe-KB.