
| Duchenne muscular dystrophy | |
|---|---|

| |
| デュシェンヌ型筋ジストロフィーの患者のふくらはぎの筋肉の断面の顕微鏡像。筋線維が広範囲にわたって脂肪細胞に置き換わっている。 | |
| 診療科 | 小児神経学, 神経筋医学, 遺伝医学 |
| 症候学 | 筋力低下、起立困難、脊椎側彎症 |
| 通常の発症 | 4歳前後 |
| 原因 | 遺伝(性染色体劣性) |
| 診断法 | 遺伝子検査 |
| 使用する医薬品 | 副腎皮質ホルモン |
| 治療 | 薬物治療、理学療法、装具療法、言語聴覚療法、作業療法、外科手術、補助換気 |
| 予後 | 平均寿命: 26歳 |
| 頻度 |
3,500-6,000人に1人(男性) 50,000,000人に1人(女性) |
デュシェンヌ型筋ジストロフィー(デュシェンヌがたきんジストロフィー、英: Duchenne muscular dystrophy、略称: DMD)は、主に男児が影響を受ける重症型の筋ジストロフィーである。通常、筋力低下は4歳前後から始まり、急速に悪化する。筋肉の喪失はまず大腿や骨盤周囲の筋肉、続いて腕でみられるようになり、その結果起立困難となる。患者の大部分は12歳までに歩行が困難になる。疾患の影響を受けた筋肉は、脂肪量の増加のために大きくなったように見える場合がある。脊椎側彎症もまた一般的である。患者の一部では知的障害も見られる可能性がある。欠陥のある遺伝子を1コピーだけ持つ女性は保因者となるが、軽度の症状がみられる場合もある。
この疾患は性染色体劣性遺伝する。疾患の原因はジストロフィンをコードする遺伝子(DMD遺伝子)の変異であり、症例の約2/3は母親からの遺伝、残りの1/3は新たに獲得された変異が原因である。ジストロフィンは筋線維の細胞膜の維持に重要な役割を果たすタンパク質である。この疾患は遺伝子検査によって出生時に診断が行われることが多い。また、患者の血液では高レベルのクレアチンキナーゼがみられる。
この疾患の根本的な治療法は知られていないが、一部の症状には理学療法、装具の使用、矯正手術が有用である場合がある。呼吸筋の筋力が低下した患者では、補助換気が必要となる場合がある。投薬としては、筋変性を遅らせるためのステロイド、痙攣や一部の筋肉の活動を制御するための抗痙攣薬、死滅しかかっている筋細胞への損傷を遅らせるための免疫抑制剤などが投与される。遺伝子治療に関してはヒトで初期段階の研究が行われており、小規模な初期研究では一部の小児で筋力の改善がみられているが、2020年の時点で長期的な影響は不明である。
DMDは3,500人から6,000人の男児に1人の割合で発生する、最も一般的なタイプの筋ジストロフィーである。平均寿命は26歳であるが、良好なケアを行うことで30代や40代まで生存する場合がある。この疾患は女児では極めて稀であり、約5000万人に1人の発生率である。
徴候と症状
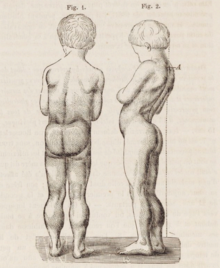
DMDでは、筋線維の錯綜、細胞死、結合組織や脂肪への置換を原因とする進行性の筋力低下が引き起こされる。まず随意筋、特に臀部、骨盤周囲、大腿、下腿の筋肉に影響が生じる。次第に肩や首、その後に腕、呼吸筋やその他の部位に進行する。疲労も一般的な症状である。
通常、疾患の徴候は5歳以前に出現し始め、歩き始めた時点からすでに観察される場合もある。運動技能は一般的に困難であり、ぎこちない歩き方や走り方となる。また、つま先歩きになる傾向があり、その一因はアキレス腱の短縮や膝伸筋の筋力不足である。転倒も頻繁に起こる。成長に伴って歩行はますます困難なものとなり、通常13歳までに歩行は完全に不可能になる。DMDの患者の男性の大部分は、21歳までに基本的に首から下が麻痺した状態となる。心筋症、特に拡張型心筋症も一般的であり、18歳の患者の約半数にみられる。うっ血性心不全や不整脈の発生は時にみられる程度である。疾患の後期段階では、呼吸障害や嚥下障害が生じ、肺炎の原因となる場合がある。
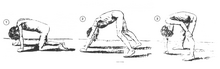
DMDの典型的な徴候は臥位または座位からの起立困難であり、ガワーズ徴候と呼ばれる特有の立ち上がり方が観察される。この立ち上がり方は子供がうつぶせの状態から起き上がろうとする際に腕を用いて骨盤周囲の筋力不足を補う方法であり、まず腕と膝で立ち、脚をよじ登るうように手を動かすことで直立する。他の特徴的な徴候には、舌、下腿、臀部、肩の筋肉の仮性肥大がある(4–5歳でみられる)。筋肉組織が最終的に脂肪や結合組織に置換される現象であるため、「仮性」肥大と呼ばれる。アキレス腱やハムストリングで筋線維の変形や拘縮が生じ、筋線維の短縮や結合組織の線維化のために機能が障害される。腰椎過前彎、脊椎側彎、骨盤前傾、胸郭変形など、骨格の変形も生じる場合がある。腰椎過前彎は、臀筋や大腿四頭筋の筋力低下に対する代償機構と考えられており、こうした変化はすべて姿勢や歩行の変化(股関節の伸展の制限など)を引き起こす。
DMDでは筋肉や骨格以外にも症状が見られる。神経行動障害(ADHDなど)、学習障害(ディスレクシアなど)、特定の認知機能(特に短期言語記憶)の非進行性の低下のリスクが高い。これらは脳内のジストロフィンの欠乏もしくは機能不全によるものであると考えられている。
DMDの発症時には、血液脳関門の破壊も特筆すべき特徴となる。
DMDの患者では血漿中のリポタンパク質濃度の上昇がみられ、原発性脂質異常症であることが示唆されている。
原因

DMDは、X染色体の短腕(Xp21)に位置するジストロフィン遺伝子の変異によって引き起こされる。変異によって、筋細胞の構造的完全性を維持しているジストロフィンタンパク質の量が大幅に減少したり、存在しなくなったりする。ジストロフィンは、各筋線維のアクチン細胞骨格を周囲の基底板(細胞外マトリックス)へ、多くのサブユニットからなるタンパク質複合体を介して連結する。ジストロフィンが存在しない場合、過剰量のカルシウムが筋鞘(細胞膜)を透過する。ミトコンドリアの機能不全によって細胞質基質でのストレス誘導性のカルシウムシグナルが増幅され、活性酸素種の産生が増幅される。いくつかの経路が関与するこの複雑なカスケードは明確には理解されていないが、細胞内の酸化ストレスの増加によって筋鞘が損傷し、最終的には細胞死が引き起こされる。筋線維は壊死し、脂肪組織や結合組織に置き換えられる。

DMDはX連鎖劣性遺伝する疾患である。患者の母親は2本のX染色体のうち1つが欠陥遺伝子であり、欠陥のあるX染色体を受け渡すことで息子に疾患が引き起こされる。男性は母親からX染色体を、父親からY染色体を受け継ぐため、保因者である母親から生まれた息子は50%の確率で疾患を発症する。保因者の娘も50%の確率で欠陥遺伝子を受け継ぐが、女性は両親からX染色体を1本ずつ受け継ぐため、父親から受け継いだ正常なX染色体が母親から受け継いだ欠陥を補償し、影響が生じることは通常はない。患者は父親としてX連鎖疾患を息子へ受け渡すことはないが、娘はその疾患の保因者となる。また、保因者の女性でも軽度の症状がみられることがある。
こうした理由により、DMDは女性では極めて稀である(約5000万出生につき1人)。父親が患者で母親が保因者である場合や、X染色体を1本欠損している場合、X染色体の不活性化(最も一般的)などによって、女性にも出現する場合がある。父親が患者で母親が保因者である場合、父親からは常に欠陥X染色体を受け継ぎ、母親からは50%の確率で欠陥X染色体を受け継ぐため、等確率で患者または保因者かとなる。
診断
DMDの疾患の家族歴がある人には遺伝カウンセリングが勧められる。DMDは妊娠時の遺伝子検査によって約95%の確度で検出することができる。
DNA検査
ジストロフィン遺伝子の筋特異的アイソフォームは79個のエクソンから構成され、DNA検査(血液検査)と解析では、影響を受けているエクソンの変異の種類が同定される。大部分の症例で、DNA検査によって確定診断となる。
筋生検
DNA検査で変異を見つけることができなかった場合には、筋生検が行われる可能性がある。生検針を用いて、少量の筋組織が採取される。DMDの生検試料に対して行われる重要な検査は、ジストロフィンに対する免疫組織化学、免疫細胞化学、イムノブロッティングによる検査であり、経験豊富な神経筋病理医によって解釈がなされるべきである。これらの検査からは、ジストロフィンタンパク質が存在するかに関する情報が得られる。タンパク質が存在しないことはDMDを意味し、存在する場合には検査によってその存在量や分子量が示されるため、DMDとより軽症のジストロフィノパチーとの鑑別に役立つ。過去数年の間に、DNA検査は疾患の原因となる変異をより多く検出できるよう進歩しており、DMDの確定のための筋生検はかつてほどは必要とされなくなっている。
出生前診断
母親が保因者であるまたは保因者であることが疑われる場合には、出生前診断が考慮される。
男児はDMDである可能性があるが、女児のDMDは極めて稀であるため、侵襲的な検査を行う前に胎児の性別を明らかにすることが重要である。性別の判定は16週で超音波検査によって行うことができ、また近年では胎児由来セルフリーDNA検査によっても行うことができる。絨毛採取(CVS)は11–14週の間に行うことができ、流産のリスクが1%ある。羊水検査は15週以降に行うことができ、流産のリスクが0.5%ある。新型出生前診断(NIPT)は11–14週前後に行うことができる。
治療
DMDの根本的治療法は存在せず、医学的需要は規制当局に認識されている。遺伝子治療の臨床試験は一部成功を収めている。
治療は一般的には症状の制御を目的としており、質問票を用いて測定されるQOLを最大化することが目的となる。次のような治療が行われる。
- プレドニゾロンやデフラザコートなどの副腎皮質ホルモン剤は、最大2年間にわたって筋強度の短期的改善をもたらす。副腎皮質ホルモン剤は歩行可能期間を延ばすことも報告されているが、そのエビデンスは強固ではない。
- 理学療法は筋肉の強度、柔軟性、機能の維持に有用である。
- 疾患の進行に伴い、適切な呼吸補助を行うことが重要である。
- 心臓の問題により、ペースメーカーが必要とある可能性がある。
モルフォリノアンチセンスオリゴ医薬品エテプリルセンは、アメリカ合衆国でジストロフィン遺伝子のエクソン51スキップに応答する患者に対して承認されている。エテプリルセンは臨床的ベネフィットが確立されなかったため、この承認には議論がある。欧州医薬品庁による承認は得られなかった。
EUでは、アタルレンの使用が承認されている。
アンチセンスオリゴヌクレオチドゴロディルセンは2019年にアメリカ合衆国で医療用途での使用が承認された。エクソン53スキップ応答症例の治療に有効である。
モルフォリノアンチセンスオリゴヌクレオチドビルトラルセンは、2020年夏にアメリカ合衆国で医療用途での使用が承認された。エクソン53スキップ応答変異が確認された患者の治療に利用される。このタイプの変異に対する標的治療としてアメリカ合衆国で2番目に承認された医薬品である。DMD患者の約8%がこの変異を抱えている。
カシメルセンは2021年2月にアメリが合衆国で医療用途での使用が承認された。エクソン45スキップ応答変異が確認された患者に対する標的治療として初めてFDAの承認を受けた医薬品である。
アメリカ疾病予防管理センターによってDMDのケアに対する多職種・包括的なガイドラインが作成され、2010年にThe Lancet Neurologyにおいて2部に分けて発表された。改訂版が2018年に発表されている。
理学療法
予後
デュシェンヌ型筋ジストロフィーは進行性の希少疾患であり、最終的には全ての随意筋が影響を受け、心筋や呼吸筋も影響を受ける。平均寿命は25–26歳と推計されているが、個人差が大きい。優れた医療ケアが行われた場合、患者は30代まで生存することが多い。この疾患で最高齢の可能性がある人物は、2021年時点で58歳であった。
DMDの患者の直接的な死因として最も一般的なのは呼吸不全である。人工呼吸器や気管切開などの治療の合併症も懸念点となる。次に多い死因は、拡張型心筋症による心不全などの心臓関連疾患である。呼吸補助が行われた場合、生存期間の中央値は40歳にまで伸びる。車いすやベッドを適切に使用し、補助換気、気道クリアランス、心臓症状に対する投薬を行うことで40代もしくは50代前半まで生存する症例も稀にある。今後のケアに関して必要なサポートを早期に計画しておくことが、DMDの患者の寿命の延長につながることが示されている。
デュシェンヌ型筋ジストロフィーのmdxマウスモデルでは、ジストロフィンの欠損がカルシウムレベルの上昇、骨格筋の筋壊死と関係している。また、内喉頭筋(ILM)は保護され、筋壊死が起こらない。ILMは他の筋肉と比較して、カルシウム濃度の変化への対処能力が高いことを示唆するカルシウム調節系プロファイルを示す。このことからはILMの独特な病態生理学的特性の根底にある機構に対する洞察がもたらされる可能性があり、またILMの研究はさまざまな臨床シナリオにおける筋消耗の予防と治療の新規戦略の開発につながる可能性がある。
疫学
DMDは最も一般的なタイプの筋ジストロフィーであり、その発生率は出生男児3600人もしくは5000人に1人と推計されている。
アメリカ合衆国の2010年の研究では、5歳から54歳までのDMDの患者の内訳は非ヒスパニック白人や黒人と比較してヒスパニックが多いことが示されている
研究
筋ジストロフィーはどのタイプも根本的治療法が存在しない。遺伝子治療(マイクロジストロフィン)やアンチセンス治療(エテプリルセンなど)といった、根本原因に対処するよう設計されたいくつかの薬剤が開発中であり、ジストロフィンまたはウトロフィン(ユートロフィン)のいずれかの産生能力を回復する薬剤の探索が行われている。他の取り組みとしては、筋細胞へのカルシウムの流入の遮断の試みなどが行われている。
また、近年の3つの進歩によって、DMDなどの筋ジストロフィーの治療状況は変化する可能性が高い。1つは、人工多能性幹細胞(iPS細胞)を用いた効果的な治療戦略のデザインである。次に、人工知能(AI)は治療標的の発見に役立つ可能性がある。また、疾患モデルを用いて多様なソースから収集された大量のマルチオミックスデータからは、さまざまな経路の収束や分岐に関して有用な情報がもたらされる可能性がある。
エクソンスキッピング
DNAの構造的アナログであるアンチセンスオリゴヌクレオチドは、DMD患者の10%の治療法となる可能性がある。これらの化合物は、ジストロフィン遺伝子がRNAへ転写される際に遺伝子の欠陥のある部分をスキップ(エクソンスキッピング)することで、切り詰められた形ではあるもののより機能的なタンパク質の産生を可能にする。
2種類のアンチセンスオリゴ、2'-O-メチルチオリン酸オリゴ(ドリサペルセン)とモルフォリノオリゴ(エテプリルセンなど)で暫定的なベネフィットのエビデンスが得られており、現在も研究が行われている。エテプリルセンはエクソン51をスキップするよう標的化されている。エクソン51のスキップによって、欠失を抱える男児の約15%でリーディングフレームが回復する。10か所の異なるエクソンを標的とするアンチセンスオリゴによって、欠失を抱える男児の70%以上への対処が可能になることが示唆されている。
DMDよりも軽症であるベッカー型筋ジストロフィーの患者では、ジストロフィンタンパク質は正常よりも短いものの、機能を保持している。1990年Englandらによって、軽症型であるベッカー型筋ジストロフィーの患者がジストロフィン遺伝子のコーディング領域の46%を欠失していることが記載された。こうした切り詰められているものの機能的な形態のジストロフィンの存在から、正常よりも短い形のジストロフィンであっても治療的ベネフィットが得られるという発想が得られた。また同時期にKoleらによって、アンチセンスオリゴヌクレオチドによるpre-mRNAの標的化によるスプライシングの変化が報告された。Koleらはβサラセミアの患者から得られた細胞で、スプライシング過程を標的としたアンチセンスオリゴヌクレオチドによって誤ったスプライシングを修正することに成功した。Wiltonのグループによって、筋ジストロフィーへのエクソンスキッピングの応用が試みられた。
遺伝子治療
DMDの原因となる変異を修正する、遺伝子編集による治療法の開発へ向けた取り組みが現在行われている。CRISPR/Cas9によるゲノム編集技術によってジストロフィン遺伝子中の変異を正確に除去し、DNA修復機構によって遺伝子の正常なコピーで置き換える、というものである。CRISPR/Cas9システムによるゲノム編集は、現在のところヒトで実行可能な状態ではない。しかしながら技術の進歩によって、将来的にはDMDの治療のためにこの技術を用いることができるようになる可能性がある。
2007年には、DMDに対するウイルスベクターを用いた遺伝子治療の最初の臨床試験が行われた。DMDやベッカー型筋ジストロフィーに対する遺伝子治療には、Biostrophinと呼ばれるデリバリーベクターが用いられている。
歴史

この疾患はナポリの医師Giovanni Semmolaによって1834年に、そしてGaetano Conteによって1836年に記載された。しかしながら、デュシェンヌ型筋ジストロフィーの名称はフランスの神経学者ギヨーム=バンジャマン=アマン・デュシェンヌ(1806–1875)に由来している。彼は1861年の著作Paraplegie hypertrophique de l'enfance de cause cerebraleにおいて、この疾患の少年の症例について詳細に記載した。その1年後、彼は自身の患者の写真をAlbum de photographies pathologiquesとして発表した。また、1868年には他の13人の患者について報告した。デュシェンヌは、顕微鏡検査のために存命中の患者から組織を採取する、生検を行った最初の人物である。
関連文献
- “Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management”. Lancet Neurol 17 (3): 251–267. (March 2018). doi:10.1016/S1474-4422(18)30024-3. PMC 5869704. PMID 29395989.
- “Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management”. Lancet Neurol 17 (4): 347–361. (April 2018). doi:10.1016/S1474-4422(18)30025-5. PMC 5889091. PMID 29395990.
外部リンク
- DMDを知る -デュシェンヌ型筋ジストロフィーの専門情報サイト-
- Muscular Dystrophies - Curlie(英語)
- CDC's National Center on Birth Defects and Developmental Disabilities (previously listed below as "Duchenne/Becker Muscular Dystrophy, NCBDDD") at CDC
- Genes and Disease Page at NCBI
| 分類 | |
|---|---|
| 外部リソース(外部リンクは英語) |