

医薬品設計(いやくひんせっけい、英: Drug design; ドラッグデザイン)とは、生物学的標的の知識に基づいて新しい薬物を見出す創意に富んだ手法である。しばしば合理的医薬品設計または単に合理的設計とも呼ばれる。薬物は、タンパク質などの生体分子の機能を活性化または阻害する有機低分子が最も一般的であり、これにより患者に治療効果をもたらす。最も基本的な意味での医薬品設計は、相互作用する生体分子標的と相補的な形状と電荷を持ち、標的に結合する分子を設計することを含む。その他にも、通常の経路を強化するために、病気の場合に影響を受けているであろう特定の分子の働きを促進する方法もある。
医薬品設計は、コンピュータ技術を用いてモデル化することができ、コンピュータ支援創薬設計とも呼ばれる。また、生体分子標的の三次元構造の知識に依存した医薬品設計は、構造ベース薬物設計として知られている。これらの技術は、活性部位の構造と性質の知見から生体分子にうまく組み合う分子の構築を可能にする。
低分子に加えて、ペプチド、特に治療用抗体を含むバイオ医薬品はますます重要で、これらのタンパク質ベースの治療薬の親和性、選択性、および安定性を向上させるための計算的手法も開発されている。
「医薬品設計」という言葉は実際には誤称であり、より正確な用語は、リガンド設計 (すなわち、標的に強固に結合する分子の設計) である。結合親和性を予測するための設計技術はかなり成功しているが、バイオアベイラビリティ、代謝半減期、副作用など、リガンドが安全で有効な薬物になる前に、まず最適化しなければならない他の多くの特性がある。これらの他の特性は、合理的設計技術では予測が難しいことが多い。それにもかかわらず、特に医薬品開発の臨床段階では離脱率が高いため、医薬品設計の初期段階では、開発中の合併症が少なく、したがって承認され、上市される可能性が高いと予測される物理化学的特性を有する候補薬を選択することに多くの関心が払われている。さらに、より良好なADME (吸収、分布、代謝、排泄) および毒性プロファイルを有する化合物を選択するために、計算法を補完するin vitro実験が創薬の初期段階で使用されるようになってきている。
薬物標的
生体分子標的(最も一般的にはタンパク質や核酸)とは、特定の疾患状態や病理学、あるいは細菌性病原体の感染性や生存性に特異的な代謝経路やシグナル伝達経路に関与する鍵となる分子である。潜在的な創薬標的は必ずしも疾患を引き起こすものではないが、定義上は疾患を修飾するものでなければならない。場合によって、低分子は、特定の疾患修飾経路における標的機能を増強または阻害するように設計される。低分子 (例えば、受容体作動薬、拮抗薬、受容体逆作動薬、または選択的受容体調節薬、酵素活性化剤または酵素阻害剤;またはイオンチャネル開口薬またはチャネルブロッカー) は、標的の結合部位に相補的であるように設計される。低分子 (薬物) は、他の重要な「オフターゲット」分子(しばしばアンチターゲットと呼ばれる)に影響を与えないように設計できる。それは、オフターゲット分子との薬物相互作用が望ましくない副作用を引き起こす可能性があるためである。結合部位の類似性により、配列相同性によって同定された密接に関連した標的は、交差反応の可能性が最も高く、それゆえに副作用の可能性が最も高いと考えられている。
最も一般的な薬物は、化学合成によって製造される有機低分子であるが、新しいアプローチとしては、生物学的プロセスによって製造される生体高分子ベースの薬物 (バイオ医薬品としても知られている) がますます一般的になってきている。さらに、mRNAベースの遺伝子サイレンシング技術は、治療への応用が期待されている。
合理的医薬品設計
これまで行われてきたような、培養細胞や動物に対する化学物質の投与による試行錯誤や、治療に対する外見上の効果を照らし合わせる、といった古典的な創薬の手法(フォワード薬理学と呼ばれる)とは異なり、合理的設計法(Rational drug discovery; リバース薬理学とも呼ばれる)は、まず体内もしくは標的器官における特定の化学反応を理解することから始まり、特定の生体分子を修飾することで治療効果が得られるのではないかという仮説を立て、これらの反応の組み合わせを治療目的に合わせて意図的に作り上げて行く。
生体分子を薬物標的として選択するためには、2つの重要な情報が必要である。第一は、標的の修飾が疾患修飾になるという証拠である。この知識は、例えば、生物学的標的の突然変異と特定の疾患状態との間の関連性を示す疾患連鎖解析から得られることがある。第二に、標的が「創薬可能性」であるということである。これは、それが低分子に結合する能力があり、その活性が低分子によって調節可能であることを意味する。
適切な標的が同定されると通常、標的はクローン化 (英語版) され、生産 (英語版) され、精製 (英語版) される。精製されたタンパク質は次に、スクリーニング試験を確立するために使用される。さらに標的の三次元構造を決定してもよい。
標的に結合する低分子の探索は、潜在的な薬物化合物のライブラリをスクリーニングすることから開始される。これは、スクリーニング試験 (「ウェットスクリーン」) を使用して行なうことができる。さらに、標的の構造が利用可能であれば、候補薬物のバーチャルスクリーンを実行してもよい。
結合部位における薬物の活性は医薬品設計の一面でしかない。さらに考慮するべき一面は分子の持つ「薬らしさ(英:druglikeness)」である。すなわち、経口バイオアベイラビリティ、適切な化学的および代謝安定性、および最小の毒性効果につながると予測される特性を備えている必要がある。薬らしさを推定する指標としてはリピンスキーの法則や、親油性効率のようないくつかのスコアリング方法が利用可能である。設計プロセス中に同時に最適化されなければならない多数の薬物特性のために、多目的最適化技術が採用されることがある。その上、化合物の代謝的安定性、安全性、さらには製造にかかる合成コストなども医薬品設計に求められる事項である。
医薬品開発はその過程の複雑さゆえに、未だにセレンディピティや限定合理性といった偶然に頼った発見を示唆する言葉が引き合いに出される。また副作用を持たない新規な医薬品となりえる化合物を、既知未知を含めた膨大な数の化学物質群から見つけ出すことは、相当なチャレンジであると言える。
コンピュータを利用した医薬品設計
医薬品設計における最も基本的な目標は、特定の分子が標的に結合するかどうか、および結合する場合にはどの程度の強さで結合するかの予測である。分子力学法や分子動力学法は、低分子とその生物学的標的との間の分子間相互作用の強さを推定するためにしばしば使用される。これらの方法は、低分子のコンホメーションを予測したり、低分子が標的に結合したときに起こるかもしれない標的の構造変化をモデル化するためにも使用される。半経験的分子軌道法、非経験的分子軌道法、または密度汎関数理論は、分子力学計算のための最適化されたパラメータを提供するためにしばしば使用され、また、結合親和性に影響を与える薬物候補の電子特性 (静電ポテンシャル、分極率など) の推定値を提供するためにも使用される。
結合親和性の半定量的な予測を提供するために、分子力学的手法を用いることもできる。また、結合親和性の予測値を提供するために、知識ベースのスコアリング関数を使用してもよい。これらの方法は、線形回帰、機械学習、ニューラルネット、または他の統計的手法を使用して、実験的親和性を低分子と標的との間の計算上導き出された相互作用エネルギーに適合させることにより、予測的な結合親和性式を導出する。
コンピュータの利用は、化合物を合成する前に親和性を予測できるため、理論的には1つの化合物のみを合成する必要があることから、理想的には膨大な時間とコストを節約することができる。しかし、現在の計算手法は不完全であり、せいぜい定性的に正確な親和性の推定値しか得られないのが現実である。実際には、最適な薬物が発見されるまでに、設計、合成、試験を数回繰り返す必要がある。計算手法は、必要な反復回数を減らすことで発見を加速し、しばしば新しい構造を提供してきた。

コンピュータを利用した医薬品設計は、次の創薬段階のいずれか使用することができる。
- バーチャルスクリーニングを用いたヒット同定 (構造ベース、またはリガンドベース設計)
- 親和性と選択性のリードジェネレーション(hit-to-lead)最適化 (構造ベース設計、QSARなど)
- 親和性を維持しつつ、他の医薬品特性のリード最適化
近代のスコアリング関数によって計算された結合親和性の不十分な予測を克服するために、タンパク質-リガンド相互作用と化合物の3次元構造情報を用いて解析が行われている。構造ベース医薬品設計では、タンパク質-リガンド相互作用に着目したポストスクリーニング解析がいくつか開発されており、エンリッチメント(富化)の向上と効率的な候補のマイニングが可能となっている。
- コンセンサス・スコアリング
- 複数のスコアリング関数の投票による候補の選択
- タンパク質-リガンド構造情報とスコアリング基準の関係を失う可能性がある
- クラスタ解析
- タンパク質-リガンドの3次元情報に基づいて候補を表現し、クラスタ化する
- タンパク質-リガンド相互作用の意味のある表現が必要。
タイプ
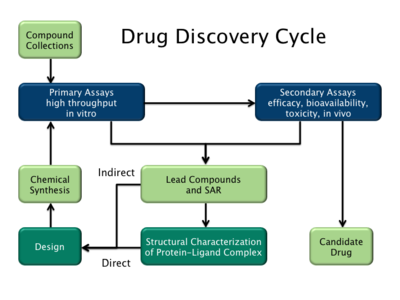
医薬品設計には大きく分けて2つのタイプがある。1つ目はリガンドベース医薬品設計と呼ばれ、2つ目は構造ベース医薬品設計と呼ばれている。
リガンドベース
リガンドベース医薬品設計 (または間接的医薬品設計) は、対象の生物学的標的に結合する他の分子の知識に依存している。このような他の分子を用いて、分子が標的に結合するために持たなければならない最低限の構造的特性を定義したファーマコフォアモデルを導出することができる。言い換えれば、生物学的標的のモデルは、それに結合するものの知識に基づいて構築することができ、このモデルを使用して、標的と相互作用する新しい分子実体を設計することができる。あるいは、計算された分子の特性と実験的に決定された生理活性との相関関係を示す定量的構造活性相関 (QSAR) を導出することもできる。これらのQSAR関係を利用して、新規アナログ(類似体)の活性を予測することができる。
構造ベース
構造ベース医薬品設計 (または直接医薬品設計) は、X線結晶構造解析やNMR分光法などの方法で得た生物学的標的の3次元構造の知識に依存している。標的の実験的構造が得られない場合は、関連するタンパク質の実験的構造に基づいて標的のホモロジーモデル(相同性モデル)を作成することも可能である。生物学的標的の構造を利用して、標的に高い親和性と選択性で結合すると予測される候補薬物を、対話的グラフィックスと薬理化学者の直感を用いて設計することができる。あるいは、様々な自動化された計算手順を用いて新薬候補を提案することもできる。
構造ベース医薬品設計のための現在の手法は、大きく3つのカテゴリーに分けられる。第一の方法は、低分子の三次元構造の大規模データベースを検索し、高速近似ドッキングプログラムを用いて受容体の結合ポケットに適合するリガンドを見つけることにより、所定の受容体に対する新規リガンドを同定する方法である。この方法はバーチャルスクリーニングとして知られている。第二のカテゴリーは、新規リガンドの新規設計である。この方法では、段階的に小片を組み立てることで、結合ポケットの制約内でリガンド分子が構築される。これらの断片は、個々の原子または分子フラグメントのいずれかである。このような方法の主な利点は、どのデータベースにも含まれていない新規な構造を提案できることである。第三の方法は、結合空洞内で提案されたアナログ(類似体)を評価することによる既知のリガンドの最適化である。
結合部位の同定
結合部位の同定は、構造ベース設計の最初のステップである。標的または十分に類似したホモログの構造が、結合したリガンドの存在下で決定される場合、リガンドは構造中で観察可能であるはずであり、その場合は結合部位の位置は些細なことである。しかしながら、関心のある未占有のアロステリック結合部位があるかもしれない。さらに、アポタンパク質 (リガンドを持たないタンパク質) の構造のみが利用可能であり、高親和性でリガンドに結合する可能性のある未占有部位を確実に同定することは極めて困難である。簡単に言えば、結合部位の同定は通常、リガンド結合を促進する適切な「ホットスポット」 (疎水性表面、水素結合部位など) も持つ薬物サイズの分子を収容することができるタンパク質上の凹面の同定に依存している。結合部位探索アルゴリズムの違いがドッキングシミュレーションプログラムの精度と関係している。
スコアリング関数
構造ベース医薬品設計は、分子認識の原理を応用して、新しいリガンドを設計するための基礎としてタンパク質の構造を利用しようとするものである。標的に選択的 (英語版) な高親和性結合は、副作用の少ない、より効果的な薬物をもたらすため一般に望ましい。したがって、潜在的な新規リガンドを設計または獲得するための最も重要な原則の一つは、特定のリガンドの標的 (および既知の抗標的(オフターゲット)) に対する結合親和性を予測し、その予測された親和性を選択の基準として使用することである。
受容体へのリガンドの結合エネルギーを記述するための、初期の汎用的な経験的スコアリング関数は、Böhmによって開発された。この経験的スコアリング関数は、次のような形式を取る。

ここで:
- ΔG0 - 経験的に導き出されたオフセット。結合時のリガンドの並進および回転エントロピーの全体的な損失に一部対応する。
- ΔGhb - 水素結合による寄与
- ΔGionic - イオン相互作用による寄与
- ΔGlip - 親油性相互作用からの寄与で、|Alipo|はリガンド-受容体間での親油性接触の表面積
- ΔGrot - リガンド結合時に回転可能なリガンドを凍結させることによるエントロピーペナルティ
より一般的な熱力学的「マスター」方程式は次のようになる。
![{\displaystyle {\begin{array}{lll}\Delta G_{\text{bind}}=-RT\ln K_{\text{d}}\\[1.3ex]K_{\text{d}}={\dfrac {[{\text{Ligand}}][{\text{Receptor}}]}{[{\text{Complex}}]}}\\[1.3ex]\Delta G_{\text{bind}}=\Delta G_{\text{desolvation}}+\Delta G_{\text{motion}}+\Delta G_{\text{configuration}}+\Delta G_{\text{interaction}}\end{array}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/ba49ddd9dec7415d129787213744ca1afcd2d021)
ここで:
- desolvation(脱溶媒) - 溶媒からリガンドを除去するためのエンタルピーペナルティ
- motion(運動) - リガンドが受容体に結合したときの自由度を低下させるエントロピーペナルティ
- configuration(構造) - リガンドを「活性」配座にするために必要な配座ひずみエネルギー
- interaction(相互作用) - リガンドとその受容体との「再溶解」のためのエンタルピー利得
基本的な考え方は、全体的な結合自由エネルギーは、結合プロセスにとって重要であることが知られている独立した成分に分解できるということである。各成分は、リガンドとその標的受容体との間の結合プロセス中に、特定の種類の自由エネルギーの変化を反映している。マスター方程式は、これらの成分の線形結合である。ギブス自由エネルギー方程式に従って、解離平衡定数Kdと自由エネルギーの成分との関係が構築された。
マスター方程式の各成分を推定するために、様々な計算手法が使用される。例えば、リガンド結合時の極表面積の変化は、脱溶媒和エネルギーを推定するために使用できる。リガンド結合時に凍結した回転可能な結合の数は、運動項に比例する。構成エネルギーやひずみエネルギーは、分子力学計算を使用して推定できる。最終的に、相互作用エネルギーは、非極性表面の変化平均力、統計的に導出された平均力ポテンシャル、形成された水素結合の数などの方法を使用して推定することができる。実際には、マスター方程式の構成要素は、多重線形回帰を用いて実験データに適合させる。これには、より正確ではないがより一般的な「グローバル」モデルを生成するために、多くのタイプのリガンドおよび受容体を含む多様なトレーニングセットを用いて行うか、または、より正確ではあるがより一般的ではない「ローカル」モデルを生成するために、リガンドおよび受容体のより限定された集合を用いて行うことができる。
医薬品設計の例
合理的な医薬品設計の典型的な例としては、X線結晶構造解析やNMRスペクトルから得られた生体分子の三次元情報の利用が挙げられる。特にコンピュータ支援創薬設計は、強力なリガンドに結合した標的タンパク質の高分解能構造が利用できる場合に、はるかに容易になる。この手法は構造を基にした医薬品設計と呼ばれることもある。実際にこの手法を用いて医薬品として承認された最初の明らかな例としては、1995年に認可を受けた炭酸脱水酵素阻害剤ドルゾラミド(Dorzolamide)が知られている。
もうひとつの合理的医薬品設計の重要な例としてイマチニブメシル酸塩(Imatinib)が挙げられる。イマチニブは慢性骨髄性白血病(CML)、フィラデルフィア染色体陽性急性リンパ性白血病)(Ph+ALL)に特徴的なBcr-Abl融合タンパクを標的としたチロシンキナーゼ阻害剤である。イマチニブがそれまでの化学療法において用いられてきた医薬品と大きく異なる点は、これまでの抗がん剤が単に分裂周期の速い細胞を標的にしていて、がん細胞と他の組織との区別するものではないのに対し、イマチニブはがん細胞とその他の正常細胞を判別することにある。
その他の例としては、以下のようなものがある。
- シメチジン (Cimetidine) - H2受容体拮抗薬で、後に開発されたヒスタミンH2受容体拮抗薬の原型となった。胃酸抑制剤。
- 多くの非定型抗精神病薬
- COX-2選択的非ステロイド性抗炎症薬 (NSAID)
- 選択的セロトニン再取り込み阻害剤、抗うつ薬
- ザナミビル (Zanamivir) - 抗ウイルス薬 インフルエンザ治療薬
- エンフビルタイド (Enfuvirtide) - ペプチド型HIV侵入阻害剤
- プロベネシド (Probenecid) - 尿酸排泄促進剤
- ゾルピデム (Zolpidem)やゾピクロン (zopiclone) などの非ベンゾジアゼピン系薬剤
- ラルテグラビル (Raltegravir) - HIVインテグラーゼ阻害剤
ケーススタディ
- 5-HT3受容体拮抗薬
- ニコチン作動薬
- アンジオテンシン受容体拮抗薬
- Bcr-Ablチロシンキナーゼ阻害薬
- カンナビノイド受容体拮抗薬
- CCR5受容体拮抗薬
- シクロオキシゲナーゼ-2阻害薬
- ジペプチジルペプチダーゼIV拮抗薬
- HIVプロテアーゼ拮抗薬
- NK1受容体拮抗薬
- 非ヌクレオシド系逆転写酵素阻害剤 (NNRTI)
- ヌクレオシドおよびヌクレオチド逆転写酵素阻害剤
- ホスホジエステラーゼ5阻害薬
- プロトンポンプ阻害薬
- レニン阻害剤
- トリプタン
- TRPV1拮抗薬
- c-Met阻害薬
批判
合理的医薬品設計の非常に厳格で焦点を絞った性質が、創薬におけるセレンディピティを抑制していると主張されてきた。最も重要な医学的発見の多くは偶然によるものであるため、最近の合理的医薬品設計への注目は、創薬の進歩を制限する可能性がある。さらに、医薬品の合理的設計は、治療を目的している疾患の基礎となる分子プロセスの未熟または不完全な理解によって制限されることがある。
関連項目
外部リンク
- Drug Design - MeSH・アメリカ国立医学図書館・生命科学用語シソーラス(英語)