
| 多発性硬化症 | |
|---|---|

| |
| 分類および外部参照情報 | |
| 診療科・ 学術分野 |
神経学 |
| ICD-10 | G35 |
| ICD-9-CM | 340 |
| OMIM | 126200 |
| DiseasesDB | 8412 |
| MedlinePlus | 000737 |
| eMedicine | neuro/228 oph/179 emerg/321 pmr/82 radio/461 |
| Patient UK | 多発性硬化症 |
| MeSH | D009103 |
| GeneReviews | |
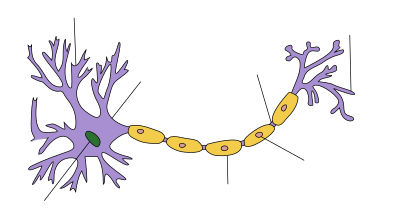
| 髄鞘をもつ末梢ニューロンの模式図。軸索にシュワン細胞が幾重にも巻き付くことによって髄鞘が形成されている。 |
|---|
多発性硬化症(たはつせいこうかしょう、英: multiple sclerosis; MS)は、中枢性脱髄疾患の一つで、神経のミエリン鞘が破壊され脳、脊髄、視神経などに病変が起こり、多様な神経症状が再発と寛解を繰り返す疾患。日本では特定疾患に認定されている指定難病である。
病名は、神経を包む組織(ミエリン鞘)が破壊されて生じる硬化が多数の領域で発生することに由来している。
疫学
地域にての発生差があり、北米、北欧、オーストラリア南部では人口10万人当たり30〜80人ほど罹患しているが、アジアやアフリカでは人口10万人当たり4人以下で、罹患率に大きな差があることが特徴である。南米、南欧、オーストラリア北部はその中間である。全体としては高緯度のほうが罹患率は高く、日本国内でも北海道と九州では北海道のほうが高い。日本での有病率は増加してきており、10万人あたり8 - 9人、人口辺り約12,000人程度であることが2006年神経免疫班会議で報告されている。
発症年齢においても罹患のピークは30歳頃であり、約80%が50歳までに発症する。また、女性に発症が多い。
原因
さまざまな説が唱えられているが未だ原因は不明である。このうち遺伝、自己免疫、ウイルス(特にエプスタイン・バール・ウイルス(EBウイルス)などの感染の可能性が高いと考えられている。
- 遺伝
- アジア・アフリカ系と欧米系で罹患率が大きく異なることから遺伝的要因が示唆されている。罹患率の高い地域に住む先住民の罹患率が高いわけではないということは遺伝説を支持する要因だが、罹患率の少ないとされる日本人やアフリカ原住民でも、有病率の高い地域に移住した場合、その発病頻度が高くなることが知られている。家族内での発症は決して高いわけではなく、複数の遺伝子が発症に関わると思われている。
- 感染
- 再発と寛解を繰り返すという病態からウイルス感染が疑われている。しかし、今まで報告されたウイルスは数多くあるものの、どれも特異的な関連ははっきり示されてはいない(最近では以下に述べるEBウイルスとの特異的な関連が示唆されている)。
- 2016年に順天堂大学らの研究者が「ヨーネ菌(MAP; Mycobacterium aviumsubsp. aratuberculosis) の関与が示唆される」とする研究成果を発表している。
- 多発性硬化症との関連が最もよく報告されているウイルスは、人間の9割が感染しているヘルペスウイルスの一種 エプスタイン・バール・ウイルス(EBウイルス)である。血清中のEBウイルス抗体価と多発性硬化症の発生リスクが強い相関を持つとする報告が多くある。しかし、多発性硬化症患者の脳ないしは脳脊髄液(cerebrospinal fluid;CSF)といった中枢神経系(central nervous system;CNS)における直接のEBウイルスの感染は稀であるという報告もあり、2011年時点では多発性硬化症とEBウイルスの直接の関連については議論の的となっていた。しかし、このEBウイルスは以下に述べるメカニズムで、自己免疫を引き起こすウイルスであると2015年に分子生物学的に証明されたことから、多発性硬化症における自己抗体産生・自己免疫応答の誘導に寄与することで多発性硬化症の病態に関わっているようである。
- 自己免疫
- 根拠は不十分であるものの、免疫異常を疑わせる所見がいくつか見られる。以下にその一例を示す。
日本をはじめとするアジア地域では、視神経と脊髄を病変の主体とする比較的症状の重い視神経脊髄型多発性硬化症が多いとされてきたが、2004年に多くの視神経脊髄型多発性硬化症の血液中に特異的な自己抗体が存在することが発見された。その後、この自己抗体はアクアポリン4(AQP4)という水チャンネルを認識することがわかり、容易に測定可能となった。現在、視神経脊髄型多発性硬化症は欧米の視神経脊髄炎(Neuromyelitis optica)と同一病態と考えられている(下記項目も参照のこと)。
大阪大学微生物病研究所/免疫学フロンティア研究センターの研究グループは2015年、自己免疫疾患の全身性エリテマトーデスや多発性硬化症との関わりが知られているEBウイルスによる自己免疫疾患発症のメカニズムを分子生物学的に示した。
通常、胚中心B細胞(成熟段階にあるB細胞)の表面に、排除する抗原に合わないB細胞受容体や、自分の抗原に反応するB細胞受容体があれば、そのB細胞はアポトーシスにより排除される。しかし、その胚中心B細胞がEBウイルスに感染すると、EBウイルスの潜伏感染III型遺伝子のLMP2AがB細胞受容体シグナルを模倣し、さらに形質細胞(抗体産生細胞)への分化を促進する因子(Zbtb20)が出現して、本来はアポトーシスにより排除されるべき自己反応性B細胞が生き残り(B細胞選択異常)、自己反応性受容体などの抗体を出し続ける形質細胞になったことから、自己免疫疾患が発症するということである。
また、クイーンズランド大学医学部の研究者は、多発性硬化症を含む自己免疫疾患のEBウイルスによる発症メカニズムを仮説の形で2003年・2011年・2012年に発表し、これは「ペンダーの仮説(Pender's hypothesis)」と呼ばれている。このペンダーの仮説は、遺伝等の原因によってEBウイルスに対するCD8+T細胞応答に何らかの不全が起き、EBウイルスに感染した自己反応性の記憶B細胞が抗原提示細胞として働き、通常は禁止された自己抗原のT細胞認識が可能となり、自己免疫応答が生ずるというものである。この仮説は2017年現在でも検証段階であるが、実際に多発性硬化症の患者においてはEBウイルスに対するT細胞応答の疲弊が起こっていること、またこの仮説により先ほどの様な中枢神経系(central nervous system;CNS)における直接のEBウイルスの感染は稀である、という様な現象を説明しうるということまで判明してきている。
エクセター大学らの研究グループによれば多発性硬化症患者はウェルシュ菌の出すイプシロン毒素に対する抗体を持っている割合が高くその関連性が指摘されている。
発症機序
多発性硬化症の発症機序と進展に関して最も広く受け入れられている仮説は自己免疫機序が一次的という仮説である。自然免疫と獲得免疫の両方が多発性硬化症の発症と進行の両方に関与している。この仮説は多発性硬化症の動物モデルである実験的自己免疫性脳脊髄炎(EAE)によって構築されている。EAEで構築された仮説は多発性硬化症の病理所見、免疫細胞の解析、免疫分子を標的とした治療によって確認されてきた。おおまかにまとめると、遺伝素因、環境要因によって自己免疫現象が起こりやすい体内環境が形成されたあと、末梢において中枢神経抗原特異的T細胞が活性化し、中枢神経系に侵入する。皮膚・消化器・呼吸器系などの感染・炎症が中枢神経抗原特異的T細胞の活性化に重要であり、T細胞の活性化は所属リンパ節で起こる。同様にB細胞の形質細胞への分化、脳内への侵入を伴うこともあり、中枢神経抗原特異的抗体を産出する。さらにT細胞由来のサイトカインなどの炎症性メディエーターはオリゴデンドロサイトやアストロサイトの構造や血液脳関門を障害し、単球、リンパ球をさらに中枢神経内に呼び込み、髄鞘を標的とした炎症により病変が検出される。 一方、オリゴデンドロサイトの異常が疾患プロセスの最初であり、中枢神経系抗原特異的免疫応答は二次的な反応であるとの仮説もある。この場合は遺伝子異常、持続的ウイルス感染などによるオリゴデンドロサイトの障害が想定される。しかし多発性硬化症において共通な遺伝子異常、持続的ウイルス感染の存在の確認はされていない。
臨床像
この節の加筆が望まれています。 |
以下の4つに分類される事もある。
- 再発寛解型
- 症状の悪化(再発)と症状の安定(寛解)が交互に起こる。数カ月から数年間の寛解期間と再発が繰り返される病態。再発原因が不明な場合もあれば、インフルエンザなどの感染症が引き金になる場合もある。
- 一次性進行型
- 病状が進行しない一時的な停滞期間があるが、寛解せず徐々に病状が進行していく。
- 二次性進行型
- 発症初期は再発と寛解が繰り返されるが、緩やかに進行していく。
- 進行再発型
- まれな病態で、病状は徐々に進行するが、突然の再発を伴う。
寛解と再発を繰り返す中枢神経系の炎症性脱髄を主として軸索変性を伴う疾患である。病変部位により症状の個人差が大きくまた同じ人でも症状の変化は大きい。中枢神経系脱髄疾患のなかで最も多く、炎症、脱髄、グリオーシスを三主徴とし寛解、再燃、進行性の経過をとる。突然健康な若年成人を主として侵す疾患であり、時に発症数週間から数ヶ月間疲労、脱力感、筋痛、関節痛がみられることもある。発症は急激なこともあれば気が付かないまま進行していることもある。初発時の発症様式は脳卒中のように数分から数時間で急激に発症する場合が20%ほどにみられる。30%で1日から数日間かけて症状が進行し、さらに20%では数週から数ヶ月間かけて症状が進行する。発症があきらかでないまま徐々に症状が進行し数ヶ月から数年にかけて慢性または間欠的に症状が進行するものもある(PPMS)。発症の誘因としては何もないことが多いが誘因として過労、ストレス、感染などが上げられている。妊娠中は再発が少なく、出産後に再発することが多い。前駆症状がない場合が多いが、時に頭痛、発熱、感冒様症状、悪心、嘔吐などが10%程度に認められる。また、過呼吸や動作時などに急に構音障害や失調症、手足のしびれや痒みなど突発性発作が現れることがある。
初発症状は脱髄病巣の部位によって多彩である。神経学的所見では無症状であると考えられた部位にも異常が認められることがある。実際に自覚症状が片側であっても、神経学的所見では両側に異常が認められることもある。四肢のしびれは初期のMSでは50%ほどに認められる。背下部の鋭い痛みは病変部位との関連は不明であるがよく認められる。日本では視力低下が最も多く、上下肢の運動麻痺、四肢頸部体感などのしびれ感がこれにつぐ。発症の状態は1〜3日で神経症状の完成する急性ないし亜急性が多い。全経過中に出現する頻度は視力低下や視神経萎縮が多い。MSでは中枢神経障害に基づく症候であればどんなものでも出現しうる。欧米に比べると日本人では急性横断性脊髄障害の頻度が高く、逆に失調症や企図振戦の頻度は低い。視神経炎が両側に起こり失明に至るような顕著な視力低下を呈する場合にはMSよりも視神経脊髄炎の可能性が高い。MSと診断された後は多くの神経症症候が定期的に生じうる。全身型のMSではおよそ半分くらいに視神経炎、脳幹、大脳、脊髄障害の症状や徴候が様々な程度呈してくる。30 - 40%位に四肢に深部異常感覚や脊髄性失調がおこる(脊髄型)。小脳型または延髄橋小脳型は5%にくらいにしかみられない。
- レルミット徴候
- 頚髄が障害された場合には頸部を他動的に前屈させると肩から背中にかけて脊柱にそって下方へ放散する電気ショック様の痛み(電撃痛)がはしる。これをレルミット徴候という。
- 視神経炎
- MSの25%に初期症状として球後性視神経炎がみられる。視力の低下、視野の異常、中心暗点が特徴的である。
- 複視
- 複視は眼筋麻痺で生じ、核間性眼筋麻痺または外転神経障害によって生じる眼球運動障害である。MSでは核間性眼球麻痺が両側性に生じるのが特徴である。このほかにMSでよくみとめられる注視麻痺には水平性注視麻痺、一眼半水平注視麻痺症候群(one and a half syndrome:水平性注視麻痺と同側の核間性眼筋麻痺)、後天性振子様眼振などがある。
- ウートフ徴候
- ウートフ(Uhthoff)徴候(またはウートフ現象)とは長時間の入浴、熱い食べ物の摂取、炎天下の外出などの結果、視力低下や筋力の低下など麻痺症状が発現あるいは悪化することである。ウートフ徴候自体が初発症状となることもある。これはすでに伝導効率が低下している傷害された神経が体温上昇に伴ってさらに伝導効率が悪化するためと考えられており、通常は冷却することで回復する。
- 急性脊髄炎(横断性脊髄炎)
- MSの場合は脊髄炎は左右非対称に生じ、不完全であることが多い。急性脊髄炎のみがみられ、その他の脱髄性病変が示唆されない場合には全身性エリテマトーデスや混合性結合組織病、抗リン脂質抗体症候群による可能性も考慮する、
- 四肢の筋力低下
- 痙縮
- 感覚障害
- Uldryらの検討では脊髄病変と感覚障害の対応は46.4%で対応があり、14.2%はおそらく対応するとしながらも全体として画像上のプラークと症候を結びつけるとは困難と報告している。特に感覚障害の分布がポリニューロパチーのパターンをとる偽多発神経炎型の存在も知られており末梢神経障害 も鑑別にあがる。背部痛や有痛性強直性痙攣(painful tonic spasm)の発作があらわれることがある。
- 小脳失調症
- 眼振、断綴性言語、企図振戦はシャルコーの三主徴として知られている。
- 膀胱直腸障害
- 認知機能障害
- 疲労
検査
MRI

診断目的の場合は造影MRIを加える事でより早期診断ができる可能性がある。無症候性Gd増強病変と非造影病変が同時に認められた場合は1回のMRIで時間的多発性 (DIT: dissemination in time) の証明ができるようになった。最初のMRIから時期を問わないフォローアップMRIにて新規T2延長病変またはGd増強病変を認めた場合もDITの証明が可能になった。空間的多発性(DIS:dissemination in space)においてもMRIは重要な役割を果たす。脳室周囲 (periventricular)、皮質近傍 (juxtacortical)、テント下 (infratentorial)、脊髄 (spinal cord) の4領域のうち2つ以上の領域においてそれぞれ1個以上のT2延長病変を認めれば空間的多発性を証明したことになる。なお、脳室周囲と皮質近傍に病変ができやすい。
MRIの撮影条件としてはテント上病変はT2WIよりもFLAIR画像の方が優れているが脳幹と基底核のMS病変はFLAIRよりもT2WIの方が優れている。MSにおけるMEI上の病変のひとつにovoid lessionがあげられる。これは楕円形の病変であり脳室に対して垂直に存在しDawson's fingerと呼ばれる。確認するにはFLAIR画像の矢状断が最も適している。病巣の活動性の評価のためしばしば造影MRIが施行される。open ring signはMSに比較的特異的とされる。MSの造影病変は4から6週間持続するが数か月持続することはなく、脳膿瘍や脳腫瘍との鑑別になる。また、造影病変はRRMSで多く見られPPMSでは少ない。T2WIで高信号を呈する病変の中にT1WIで低信号を示すものがありblack holeとよばれる。視神経炎を疑うときに冠状断MRIで脂肪抑制T2WIで高信号に視神経が描出されることがある。視神経炎の活動性評価のために脂肪抑制GdT1WIを撮影することもある。MRSもよく用いられる。また、MSを疑うときは脳MRIだけではなく全脊髄MRIも撮影する。神経症状の増悪を認めなくとも定期的なMRI撮影が必要である。画像上病変の増加が認められることがある。
注意するべきこととしてMRIで異常が認められなくともMSの再発は否定できない。髄液検査でも異常が見られないこともあり、症状から再発が強く疑われたときは画像所見、髄液所見の結果に関係なくステロイドパルスを思考するべきという意見もある。
髄液検査
特異的な髄液中のマーカーは見つかっていない。髄液細胞数や蛋白は正常なことが多く、上昇しても軽度である。細胞数が極端に多い場合はむしろ他の疾患を考慮する。特に好中球が優位な場合は視神経脊髄炎が検討される。OCBやIgG indexは現在MS診断において最も用いられている髄液検査でありそれぞれ髄腔内でのIgG産出を質的、量的に評価するものである。欧米の報告ではMSのOCB陽性率は95%とされるが日本人では70%程度とされ陰性例の判断にも注意が必要である。OCB陽性例はCISならばMSへの移行率が高く、MSならば障害度の進行が早く予後予測の点でも重要である。MBPの測定もよく行われる。
- オリゴクローナルバンド (OCB)
- OCBは髄液を電気泳動し、免疫グロブリンを特異的に染色した際にγグロブリン領域に細く濃染する数本のバンドのことである。オリゴクローナルバンドの存在はある抗原に対して、とくに強い液性免疫応答が起こっていることを示している。特に同時採血した血清中に対応するオリゴクローナルバンドがなく、髄液で認められれば中枢神経内での抗体産出を意味する。オリゴクローナルバンドは脱髄疾患、感染症、末梢神経障害などで陽性となる。脱髄疾患には多発性硬化症や副腎白質ジストロフィー、感染症では各種髄膜炎、神経梅毒、亜急性硬化性全脳炎、進行性多巣性白質脳症、HAM、HIV-1感染症などで知られている。また、脳血管障害やSLE、脳膿瘍などでも認められる。膠原病や梅毒、亜急性硬化性全脳炎などの感染症などの場合でオリゴクローナルバンド陽性の時は治療とともに消失していくのが特徴とされている。
- ミエリン塩基性タンパク質 (MBP)
- MBPはミエリンを構成する主要タンパク質である。MBPの上昇は髄鞘の破壊の亢進を意味する。MBPが高値になる疾患としては、多発性硬化症、亜急性硬化性全脳炎(SSPE)、神経梅毒、脳炎各種、神経ベーチェット病、ギランバレ症候群、慢性脱髄性多発神経炎(CIDP)、HAM、頭部外傷、脳梗塞急性期、AIDS dementia complexなどが知られている。
神経生理検査
MSではMRIで描出されない潜在性病変の検出に誘発電位検査が有用である。視覚誘発電位、体性感覚誘発電位、運動誘発電位が用いられる。複数の誘発電位検査を組み合わせることでMS病変の空間的多発性の証明に役立つ。
自己抗体
病型分類評価のためにも以下の抗体測定は必須となっている。
- 抗AQP4(aquaporin-4)抗体
-
視神経脊髄炎(NMO)関連疾患(NMOSD)の診断に必須である。
- 視神経脊髄炎(neuromyelitis optica;NMO)は我が国に多く、以前はMSの亜型(視神経脊髄型)と考えられていたが、血清の抗アクアポリン4抗体が陽性となることが発見され、現在では別疾患として扱われている。NMOはMSと同様に女性に多いが、発症年齢がより高い。脳脊髄液検査では細胞と蛋白の増加が比較的高度であるが、オリゴクローナルバンドの陽性率は低い。
- 抗MOG(myelin-oligodendrocyte glycoprotein)抗体
- 鑑別目的として施行され、通常のELISA法では困難でCBA法で測定される。陽性であれば急性散在性脳脊髄炎(ADEM)やの可能性を考慮していく。
診断
中枢性脱髄疾患を臨床上/画像検査上考慮した場合には、以下の3つの状態を診断していく。
CIS
多発性硬化症(MS)患者の多くは、最初は単一の臨床症状を呈していることが知られており、これを「clinically isolated syndrome(CIS)」と言う。神経の1箇所以上の部位の炎症性脱髄病変により引き起こされた24時間以上持続する初回の神経症候であり、通常は1箇所の中枢神経病変であり視神経炎による右眼視力低下などであるが2箇所以上の中枢神経病変が同時におき視神経炎と片麻痺が同時に起こることもある。その後、初発時と異なる病変に起因する神経症候が生じ、再発と判断されるとその段階で臨床的に時間的多発性および空間的多発性が確認されたことになり、臨床的に診断確実なMS(CDMS:clinically definite MS)となる。CISの時点で1個以上MS様病変あれば長期的には80%以上の症例が再発して臨床的に診断確実なMSとなる。CISの時点で全くMS様病変がない場合はMSへの移行は20%程度と報告されている。また、CISの時点で脳MRIのT2延長病変が多いほど発症から20年後に歩行に補助を要するEDSS6.0に達する可能性が高くなる。CISに関してはMSの鑑別疾患に関する国際委員会の分類が有名である。type 5 CISはRIS(radiologically isolated syndorome)という。
また、臨床症状ではなく人間ドック等においての頭部MRI検査の画像検査で偶然に認めるものを「Radiologically isolated syndrome(RIS)」と言う。
| 分類 | 内容 | MSへの移行率 |
|---|---|---|
| type 1 CIS | 臨床的にmonofocalで1個以上の無症候性MRI病変あり | 高い |
| type 2 CIS | 臨床的にmultifocalで1個以上の無症候性MRI病変あり | 高い |
| type 3 CIS | 臨床的にmonofocalで無症候性MRI病変なし | 比較的低い |
| type 4 CIS | 臨床的にmultifocalで無症候性MRIなし | まれ |
| type 5 CIS | 脱髄性疾患を示唆する臨床症候はないがMRI所見はMSを示唆する | 不明 |
| 部位 | MSでよくみられるCISの特徴 | MSで見られることもあるが頻度の低いCISの特徴 | MSではほとんど見られない非典型的なCISの特徴 |
|---|---|---|---|
| 視神経 | 一過性視神経炎、眼球運動に伴う眼痛、部分的あるいは主に中枢性の視覚障害、正常の視神経乳頭または軽度の視神経乳頭浮腫 | 両側同時発症の視神経炎、眼痛なし、無光覚、出血を伴わない中等度または重度の視神経乳頭腫脹、ぶどう膜炎(軽度、後部) | 進行性視神経症、重度の持続性眼窩部痛、持続性の完全失明、神経網膜炎(macular starを伴う視神経乳頭浮腫)、ぶどう膜炎(重度、前部) |
| 脳幹/小脳 | 両側核間性眼筋麻痺、小脳失調および複数の眼位でみられる眼振、外転神経麻痺、顔面の感覚低下 | 一側性核間性麻痺、顔面麻痺、顔面ミオキミア、難聴、一眼半水平注視麻痺症候群、三叉神経痛、発作性緊張性痙攣 | 完全外眼筋麻痺、垂直注視性麻痺、血管領域症候群、動眼神経麻痺、進行性三叉神経感覚障害、限局性ジストニア、斜頚 |
| 脊髄 | 非横断性脊髄症、レルミット徴候、求心路遮断された手、感覚低下、尿意切迫、尿失禁、勃起不全、非対称性進行性痙性対麻痺 | 完全横断性脊髄症、神経根症、反射消失、髄節性温痛覚消失、部分的ブラウンセカール症候群(後索障害なし)、便失禁、対称性の進行性痙性対麻痺 | 前脊髄動脈領域病変(後索のみ障害なし)、馬尾症候群、境界明瞭な全感覚の感覚レベルと限局性脊髄性疼痛、完全なブラウンセカール症候群、急性尿閉、進行性感覚失調(後索) |
| 大脳半球 | 軽度の皮質下性認知機能障害、不全片麻痺 | てんかん、半盲 | 脳症(鈍麻、錯乱、傾眠)、皮質盲 |
MSの病勢は発症早期はむしろ活発である。MSの発症早期には臨床症状が比較的軽症であるが病勢は高く、治療を遅らせるのは適切ではない。CISの時点で治療開始が望ましい。慢性進行型になると血液脳関門の破綻が就職され薬剤が到達しにくくなること、神経変性の要素が病態に加わり免疫学的治療薬の有効性が乏しくなることから早期介入が望まれる。
MS
多発性硬化症(MS)は臨床症候やMRIによって炎症性脱髄によると判断される病変が時間的多発性(DIT:dissemination in time)と空間的多発性(DIS:dissemination in space)を呈する。急性増悪を繰り返す再発寛解型(RRMS)と発症時から急性増悪がなく1年以上にわたり徐々に病状が進行していく一次進行型(PPMS)はMcDonald基準によりMRI所見や髄液所見を考慮して高い精度で早期診断がすることが可能である。RRMSとして経過した後に慢性進行型に移行するSPMSはMcDonald診断基準では定義されていない。
診断基準は、1954年のAllisonの基準、1965年のSchumacherの基準、1983年のPoserの基準、1988年の旧厚労省の基準、2001年のMcDonald基準が知られており、McDonaldの診断基準は2005年と2010年に改訂がされているが、現在基本的にこの「McDonaldの診断基準」が広く用いられている。McDonald基準は初版から変わっていない基本原則が4つあり、1つはMcDonald基準は中枢神経病変のDITとDISを証明するための基準であるということ。発作(増悪、再発)には定義がある。それは中枢神経症候が炎症性脱髄によると考えられ、患者の主観的な報告あるいは客観的な観察によるものであり24時間以上持続しpseudo-relapseや再発性でない突発性症候が除外されており、ある発作の発症と次の発作の発症の間隔は30日以上であることが必要である。なお、病歴上の神経症状であって現在はその症状が見られない場合はMRIでそれに関連する病変の有無を検証する必要がある。診断はMS、possible MS(CIS)、non MSとなるということである。
自然経過から多発性硬化症は以下に大別される。
- 再発寛解型 (relapseing-remitting MS:RRMS)
- 再発寛解を繰り返すもの。ほとんど初期の場合の多くはこの症状を呈する。
- 進行型
-
- 一次性進行型 (primary-progressive MS:PPMS)
- 発症当初から進行性の経過を経るもの。
- 二次性進行型 (secondary-progressive MS:SPMS)
- RRMSの中で発症後15〜20年の経過で再発がなくても次第に障害が進行するようになるもので、再発は炎症過程を示しており進行は変性過程を示していると考えられている。非常に治療抵抗性で治療コントロールが困難。
欧米白人ではRRMSが80〜90%であり、PPMSが10〜20%を占めるが日本人ではPPMSは5%前後である。RRMSとPPMSは治療に対する反応性の違いから異なる疾患とする立場と、長時間の自然経過の観察に基いてRRMSもPPMSも同じような年齢で同様な障害度に進行することから、1つの疾患の異なる表現型とする立場がある。EDSSスコアで4に達するまでの期間(進行のスピード)は病型によって異なるがスコア4からスコア6に至る期間は病型は再発の有無に関係なく一定である。スコア6にはPPMSでは49歳、RRMS/SPMSでは48歳であり、スコア8に達するのはともに58歳である。進行型の病態には神経変性が関わると考えられている 。
| RRMS | PPMS | |
|---|---|---|
| MSに占める頻度 | 85〜90% | 10〜15% |
| 性比(男:女) | 1:2〜3 | 1:1 |
| 平均発症年齢 | 30歳 | 40歳 |
| 主たる症候 | 脊髄(感覚優位)、視神経、脳幹症候 | 痙性対麻痺、小脳性運動失調 |
| 脳MRI上のGd造影病変 | よくあり | 少ない |
| 早期の脊髄萎縮 | まれ | あり |
| 髄液OBの頻度 | 90% | 80% |
| 経過(車椅子生活までの期間の中央値) | より遅い(33.1年) | より早い(13.4年) |
| IFNβ治療効果 | あり | なし |
MSの障害は年齢に依存し、RRMSでは初回発作からの完全回復率は高齢者では若年者より有意に低下する。MSの予後不良因子としては男性、高齢発症、PPMS、初発の運動症候、小脳症候、膀胱直腸障害の存在、再発間隔の短さ(年間再発率の高さ)、病初期の再発の多さ、初期からの障害の残存、より多くの神経機能障害、発症5年後の障害度の高さとMRI lession loadの多さがあげられる。
- 良性型MS (benign MS)
- 良性型MSの定義は一様ではないももの発症5〜10年後にEDSSスコア3点以下のものは10〜20年後に障害を呈するリスクが極めて低いといわれている。しかし発症10年後にEDSS3.0以下であっても約半数が認知機能障害をおこすといった報告や発症10年後にEDSS3.0点以下であっても20年後にEDSSが3.0以下のものは半数であり、20%はEDSS6.0以上となり歩行に補助が必要なレベルという報告もある。経過は必ずしも軽症のまま推移するとは限らない。
- 急性(劇症型)MS
- 大脳、脳幹、脊髄などの多彩な症状が2〜3週間のうちに出現し昏睡など顕著な意識障害をきたし数週間から数ヶ月のうちに寛解をみることなく死に至る、劇症の経過をとるMSである。剖検例では急性散在性脳脊髄炎と異なり比較的大きな典型的なMSの肉眼的脱髄斑が多数認められる。アフェレーシスが有効なことが多く救命例の報告が増えてきつつある。
NMO
視神経脊髄炎(neuromyelitis optica:NMO)はかつて多発性硬化症の亜型(視神経脊髄型)として考えられていた疾患であり、特徴として女性に多く、発症年齢が比較的高く、髄液細胞と蛋白の増加が比較的高度であるがオリゴクローナルバンドの陽性率は低い。頭部MRI所見が軽微、脊髄MRI所見が高度、高カルジオリピン抗体やMPO-ANCAなど自己抗体の発現頻度が高い。内分泌異常を伴いやすいという特徴がある。検査上抗aquaporin-4(AQP4)抗体陽性が特異的で(多発性硬化症では陽性にならない)、多発性硬化症よりも失明に至るような重篤な視神経炎を起こしやすいが、急性期の血漿交換療法(血液浄化療法)が有効である。このように、多発性硬化症とは異なる特徴が多いことから、現在では別疾患として扱われている。
診断基準は、1999年のWingerchuk基準と、2006年改訂の同基準が広く用いられている。
病理
多発性硬化症は中枢神経系の髄鞘を標的とする炎症性脱髄性疾患である。一般的な病理像としてはT細胞主体の炎症細胞浸潤、脱髄、グリオーシス、種々の程度の軸索障害、髄鞘再生を特徴とする。炎症および変性機構による皮質脱髄と認知機能障害に関しても注目されている。
急性期
急性期には病巣内に活性化マクロファージ/ミクログリア、T細胞を主とした炎症細胞が充満する。これをhypercellularityという。T細胞は病巣内の血管周囲に集簇する。これをperivascular cuffという。同時にT細胞は脱髄巣全体にも広く認められる。浸潤したT細胞数は活性化マクロファージ/ミクログリアよりも少ない。脱髄病巣内で泡沫状マクロファージに髄鞘崩壊産物の貪食像が確認された場合には活動性脱髄(active demyelination)と表現される。髄鞘蛋白の中でもmyelin-associated glycoprotein(MAG)やmyelin oligodendrocyte glycoprotein(MOG)やcyclic nucleotide disphoesterase(CNS)などsmall molecular weight myelin proteinの貪食像が確認できるのが約2日(early active demyelination)、myelin basic protein(MBP)やproteolipid protein(PLP)のようなmajor myelin proteinの貪食像は約6~8日、KB染色では約10日(late active demyelination)と確認される時期は異なる。また、脂質染色であるSudan染色陽性マクロファージは数カ月間存在する(inactive demyelination)。S100タンパクファミリーに属するMRP14免疫染色も多発性硬化症の病期分類に有効でありactive and early demyelinating lesionにおける活性化マクロファージ/ミクログリアで発現が確認できる。オリゴデンドロサイトは顕著に脱落するという報告や、一方では数が保たれるという報告もある。炎症の活動期には血液脳関門の障害を伴うこともある。これは頭部MRIでのGd増強効果で判断される。完成された脱髄病巣では、様々な程度の軸索障害を合併し、一部は髄鞘再生の所見も認められる。これら局所的な炎症性脱髄は罹病期間の短い多発性硬化症や再発寛解型多発性硬化症で多く認められる。二次進行型多発性硬化症でも認められるが罹患期間とともに頻度は減少する。超急性期病変ではリンパ球の浸潤前にオリゴデンドロサイトの消失とミクログリアの活性化が始まっているという報告もあり、免疫応答が開始される前に組織破壊が始まるという仮説もある。
Lucchinettiらは多発性硬化症の初期病巣を4パターンに分類した。パターンIはT細胞やマクロファージの浸潤が主体であり、髄鞘再生を示すshadow plaqueが認められる。パターンIIはパターンIに加えて免疫グロブリンや補体の沈着を伴い、B細胞や形質細胞の浸潤も認められる。shadow plaqueも認められる。パターンIIIとパターンIVはオリゴデンドロサイトの脱落を主体とするものでprimary oligodendrocyte dystrophyが示唆されている。パターンIIIは髄鞘の最内層に発現するMAGやCNPの脱落が先行し、オリゴデンドロサイトのアポトーシス様変化に伴うdistal oligodendrogliopathy (DO)型脱髄が大きな特徴である。さらにパターンIIIの一部ではバロー病様の同心円状病巣が認められる。パターンⅣは一次進行型多発性硬化症に例外的い認められ、DNA断片化を伴うオリゴデンドロサイトの脱落が際立つ点で区別される。これらによりパターンI、パターンIIはT細胞による細胞性免疫が主体で髄鞘を標的とした脱髄であり、パターンIIIとパターンIVはオリゴデンドロサイトが標的となりウイルス感染や毒素の関与が歌われる。さらにLucchinettiらは多発性硬化症の病理所見の経時的比較し、22例中21例(95%)で同一パターンの病理所見が得られたと報告した。これは一個体内での病理学的均一性(intraindividual pathological homogeneity)および個体間での不均一性(interindividual heterogeneity)を支持する。その一方でBarnettらはすべての多発性硬化症の病巣の極初期にはパターンIIIのオリゴデンドロサイトのアポトーシス様変化が認められ、その後パターンII様の脱髄変化と補体の活性化がみられたことを報告しており、病理学的な不均一性は個体ごとではなく病期によるものと結論付けている。
慢性期
慢性期の病巣は境界明瞭で髄鞘は完全に脱落し、髄鞘貪食マクロファージは認められない。全体に細胞成分は減少しているが、病巣内の血管周囲性にリンパ球やマクロファージが残存する。反応性アストロサイトによる線維性グリオーシスも顕著に認められる。軸索は様々な程度で脱落しており、成熟オリゴデンドロサイトは認められない。
進行型多発性硬化症
一次進行型多発性硬化症の病理像はオリゴデンドロサイトの変性が主体で、炎症は目立たないがT細胞浸潤を長期に認め、活動性の軸索障害は少ない。二次進行型多発性硬化症では局所的な活動性脱髄病巣は比較的まれで、髄鞘貪食マクロファージや血管周囲性細胞浸潤は乏しい。病巣辺縁には活性化ミクログリア、活性化補体の沈着、髄鞘破壊の進行が認められる。完成した古い病巣でも進行性の脱髄が得られることが特徴とされている。通常の頭部MRIでは異常が認められない白質をnormal appearing white matter(NAWM)という。進行型多発性硬化症のNAWMでは活性化ミクログリアやT細胞を主とした炎症細胞浸潤や軸索障害が認められるが、活動性脱髄初見に乏しい。さらに進行型多発性硬化症では広範な皮質脱髄を認めることがある。皮質脱髄は主に大脳の軟膜下層で多く、軟膜に単核球の炎症性浸潤を伴うことがある。
皮質脱髄、髄膜炎症、異所性リンパ濾胞様構造
多発性硬化症の皮質病巣では以前からグルタミン酸トランスポーターEAAT2の脱落が報告されている。グルタミン酸毒性による組織障害が示唆されている。それとは別に、多発性硬化症における皮質脱髄や髄膜炎性が報告され、認知機能障害への関与が示唆されている。2011年にLucchinettiらは多数の早期MS患者の生検標本や剖検例を用いて炎症性皮質脱髄が生じていることを明らかにした。皮質脱髄は軟膜下、皮質内、皮質白質境界部のいずれでも観察され、髄膜炎症と局所的に関連していることも見出した。さらに一個体標本で軟膜下と皮質白質境界部ともに脱髄を認めた症例から、サイトカインなど液性因子の拡散が深部病巣形成に寄与している可能性も示唆された。一方、二次進行性多発性硬化症患者の軟膜にB細胞主体の異所性リンパ濾胞様構造(ectopic lymphoid follicle-like structure)が確認され、免疫細胞の活性化が慢性期における病変拡大、髄膜炎症や皮質脱髄との関連が示唆されている。異所性リンパ濾胞様構造の病態生理における役割は明らかではないが、向炎症性メディエーターや自己抗体、自己反応性T細胞などの局所における供給源となっている可能性がある。
オリゴデンドログリアサイト
急性期病巣ではオリゴデンドロサイトは顕著に脱落するという報告や、一方では数が保たれるという報告もある。個々の症例や病期によって組織障害の差がみられることが推測される。
アストロサイト
アストロサイトはシナプスやランビエ絞輪、血液脳関門と足突起を介して連絡を可能とするグリア細胞でありコネキシンが構成するギャップジャンクションを介してアストロサイト間やアストロサイト-オリゴデンドロサイト/ミエリン間の機能連絡も行っている。多発性硬化症の急性期脱髄病巣ではHE染色で赤く肥大した胞体を有するアストロサイトが多数認められ肥大アストロサイトと呼ばれる。肥大アストロサイトは中間径フィラメントであるGFAPとvimentin、nestinの発現が亢進する。また、一部の肥大アストロサイトは特に膨化が著明で核が細かい青色の顆粒状に認められ有糸分裂を想像させる。このアストロサイトは特にクロイツフェルト細胞と表現される。急性期の脱髄病巣に比較的特徴的な所見とされ、治療前の脳生検組織などでも観察されることがある。急性期脱髄病巣ではAQP4やconexin 43(Cx43)が広範に脱落する。アストロサイト足突起の機能タンパクが早期から脱落することが病態へ寄与している可能性がある。
軸索障害
かつては多発性硬化症は軸索が保たれるのが特徴とされていた。しかし活動性脱髄病巣でAPP染色では軸索障害が報告されている。
髄鞘再生
髄鞘再生は長く議論されているが、実験動物や電子顕微鏡研究からその存在が証明されている。髄鞘再生は脱髄病変の辺縁/境界部でよく観察されるが病巣中心部でも起こりうるとされ、斑状にもびまん性にも生じうる。現状では障害のない髄鞘と再生した髄鞘を明確に区別できる分子マーカーは存在しない。病巣内に広範な髄鞘再生が生じると病理学的にはshadow plaqueと表現されLFB染色で淡く染色される境界明瞭な病巣となり、薄い髄鞘を有した軸索が認められる。shadow plaqueは多発性硬化症の急性期でよく認められ、オリゴデンドロサイト前駆細胞(oligodendrocyte precursor cell、OPC)が動員され分化増殖することで髄鞘再生が行われる。多発性硬化症の病巣のOPCには正常では発現していないTIP30が発現し、転写因子など核移行物質の輸送を阻害している可能性が報告され、多発性硬化症におけるOPC機能異常が示唆されている。OPCは血液脳関門にも存在し脳血管に沿って遊走することも報告されている。さらに血液脳関門を構成するペリサイトなどと相互作用する可能性も報告されている
経過
RRMSの標準的な自然経過をまとめる。MSになりやすい素因は免疫系が形成される15歳までに獲得される。平均して30歳ころに臨床的に明らかな初回発作を起こし、再発、寛解を繰り返す。初発時にすでに複数の潜在的な脳MEI病巣を有することが多い。臨床的な発症に先行して潜在的な病巣の形成は多くの患者で生じている。再発は中枢神経系のどこでも生じうる。急性脱髄性炎症に伴う軸索の切断は早い時期から生じていることが病理学的に証明されている。中枢神経の可塑性や再髄鞘化により再発は病初期は回復しやすい。しかし次第に軸索障害が蓄積することにより再発後に後遺症を残すようになる。再発頻度は発症後数年が最も効率であり経過が長くなるにつれ年間再発率は自然に減少する。発症後15〜20年の経過で再発がなくても障害が次第に進行するようになり二次性進行期にはいる。二次性進行期では進行性の障害をきたす病巣は中枢神経のどこでもおきるわけではなく、錐体路の遠位部に生じやすく痙性対麻痺が悪化していく形をとりやすい。ついで小脳が障害されやすく小脳性運動失調が次第に増悪する。二次性進行期はEDSS3.0レベルからすでにはじまっていると考えられている。平均寿命は一般人と同じ程度か10年ほど短縮する。死亡率も同年齢の一般人口より3倍程度高いが1950年以降、死亡率の増加は軽減されている。
長期予後を改善する病態修飾薬(disease modifying drug:DMD)の登場で機能予後も改善されつつある。
治療
CIS/MS(多発性硬化症)/NMO(視神経脊髄炎)の診断の後に、治療としては以下を行っていく。
急性増悪期
- 副腎ステロイド薬 (corticosteroid:CS)
- CIS/MS/NMOいずれにおいても、急性期増悪期はステロイドパルス療法が推奨される。症状の改善が悪いときや重症の再発の場合は後療法としてプレドニゾロンを0.5〜1.0mg/kg/dayの投与を行い2〜3週間で漸減中止する。MSでは経口副腎ステロイド内服に再発予防効果はないが視神経脊髄炎(NMO)では経口副腎ステロイド内服と免疫抑制剤の併用が再発予防に有効と考えられている。定期的ステロイドパルス療法は多発性硬化症の脳萎縮の進行の抑制に有効である可能性がある。
- 血漿浄化療法 (plasmapheresis:PP)/血漿交換療法(plasma exchange:PE)
- CIS/MS/NMOいずれにおいても、ステロイドパルス療法が無効なときは血液浄化療法(アフェレーシス)を検討する。ステロイド治療の効果が十分でない症例において早期から思考するべき治療で、基本的に単純血漿交換療法(sPE)が行われる。多くはRRMSの急性増悪期の治療として用いられ、PPMSでは適応はない。
再発・進行防止
CIS/MS/NMOにおいて、その後の再発進行予防として、以下の治療を行っていく。
長期予後を改善する薬剤という意味で、「病態修飾薬(DMD)」と呼び、開発経緯からインターフェロンβがはじめてのDMDで、その後開発に合わせて第一世代DMDや第二世代DMDと呼ばれている。
インターフェロンβ (IFNβ)
インターフェロンβはTh1型の免疫応答をTh1抑制するTh2型へ偏倚させる作用(Th2シフトとよばれる)によって再発予防効果をもたらすと考えられている。しかしTh2細胞の産出するIL-4やIL-5は抗体産出の方向へ免疫応答を促進させるため自己抗体が関与する膠原病合併患者ではIFNβの積極的使用は推奨されていない。IFNβは糖鎖の有無によってIFNβ-1b(ベタフェロン、隔日皮下注)とIFNβ-1a(アボネックス、週1回筋注)がある。IFNβはMSの再発を予防し、身体機能障害の進行を抑制することが期待される。RRMSが最もよい適応であるが二次性進行性MSであっても臨床的あるいは画像上の再発を認める場合には治療効果がきたいできる。投与開始が早期であるほど、投与期間が長期であるほど高い治療効果が期待できる。RRMSならば再発率を約30%も低下させ、脳MRI上活動性病巣の出現も60〜80%抑制し、臨床的に中等度以上の再発を約50%低下させる効果も示されている。CISに対してはIFNβ-1aでは2年以内のCDMSへの進展する割合が偽薬群で38.6%でありIFNβ-1a群では21.1%であり相対リスクを44%低下(p=0.002)させた(CHAMPS試験)。IFNβ製剤では容量に関しては一般的に天井効果があると知られている。有効性に製剤間で差がないとする報告が多い一方、高用量で高頻度のIFNβがより有効との報告もある。副作用としてはインフルエンザい様症状、注射部位反応、うつ状態、臨床検査値異常(白血球減少、リンパ球減少、肝機能障害が多い)、月経異常などが知られている。インフルエンザ様症状に関してはNSAIDsが有効で無効時は経口ステロイドを考慮する。検査値異常は投与開始後6ヶ月以内に出現し時間の経過とともに安定することがほとんどである。用量依存性であり少量より開始し有害事象の発現をみながら漸減することが推奨される。投与開始後1ヶ月は1〜2週間ごとにその後は1〜2ヶ月毎に血液検査を行うことが望ましい。自己抗体や甲状腺機能は3〜6ヶ月毎の検査が望ましい。IFNβ治療は妊娠中は禁忌であり避妊が必要である。また、小紫胡湯の併用で間質性肺炎がおこることがある。
IFNβ製剤は蛋白製剤であるため、IFNβに対する免疫応答の結果、中和抗体が出現することがある。出現頻度は製剤の種類や投与経路、投与間隔によって異なり、一般的にはIFNβ-1aよりもIFNβ-1b、筋注よりも皮下注、投与間隔が短いほど出現頻度が高い。IFNβで再発予防効果が乏しい時は中和抗体の影響を考慮する必要がある。
免疫抑制剤
免疫抑制剤では以下の治療薬が用いられる。旧来よりIFNβ抵抗性症例に用いられるが、ほとんど保険適応未。
- グラチラマー(コパキソン)
- RRMSにおける再発予防として用いられる。進行型への効果は示せていない。
- フィンゴリモド(イムセラ、ジレニア)
- はじめて経口内服で再発防止作用を発揮する薬剤である。フィンゴリモドは多発性硬化症患者の末梢を循環している自己反応性Tリンパ球をリンパ節内にとどめることで、その中枢神経への浸潤を抑制し中枢神経における炎症をおさえる。RRMSにおける再発予防効果はIFNβより高いとされている。進行型MSやNMO、3椎体以上の脊髄病変のある例に対する有効性は確立していない。また、有害事象として免疫抑制でのJCウイルス活性による進行性多巣性白質脳症(progressive multifocal leukoencephalopathy:PML)の出現があることが知られている。日本国内においては少なくとも4名の患者がフィンゴリモド投与中にPMLを発症したことが確認されている。PMLの発症に気付くのが遅れた場合は重症化から死亡に至るケースもあるため、MRIによる経過観察が必要である。また、投与にあたっては慎重に検討することが望ましい。
- ミトキサントロン(ノバントロン)
- 保険適応未。ただ旧来よりMSに対して用いられている。
- メトトレキサート(メソトレキセート)
- 保険適応未。ただ旧来よりMSに対して用いられている。
- アザチオプリン(イムラン アサニン)
- 保険適応未。ただ旧来よりNMOに対して用いられている。
- シクロホスファミド(エンドキサン)
- 保険適応未。
- テリフルノミド
- レフルノミドの活性代謝物で日本では保険申請中。
- フマル酸ジメチル(テクフィデラ)
- 経口内服として投与され、RRMSに対して高い再発防止効果と症状進行抑制効果を示している。有害事象としてフィンゴリモド同様にJCウイルス活性による進行性多巣性白質脳症(progressive multifocal leukoencephalopathy:PML)の出現があることが知られており、慎重に投与検討される。テクフィデラ投与中のPML出現例では、長期にわたりリンパ球数の減少がみられるケースが多い。そのため、投与中には定期的にリンパ球数のモニタリングを行うことが推奨されている。
分子標的治療薬
IFNβや免疫抑制剤でも再発進行を生じる治療抵抗性症例に以下の分子標的治療薬薬が用いられる。
- ナタリズマブ(タイサブリ)
- ナタリズマブはヒト化抗α4インテグリン・モノクローナル抗体で、治療抵抗性の多発性硬化症(MS)に対して非常に強い再発防止効果と症状進行抑制効果を示している。ただ有害事象として免疫抑制でのJCウイルス活性による進行性多巣性白質脳症(progressive multifocal leukoencephalopathy:PML)の出現があることが知られている。JCウイルスに対する抗体が陽性の場合、投与から1年を超えると出現リスクが上昇するため、慎重に投与検討される。
- アレムツズマブ(マブキャンパス)
- 保険適応未。
- リツキシマブ
- 抗CD20抗体, 保険適応未。
- オファツムマブ (ケシンプタ)
- 抗CD20抗体
- ダクリズマブ
- オピシヌマブ
- 抗LINGO-1抗体で、急性視神経炎患者に対して改善効果が報告。
- トレブルチニブ
- ブルトン型チロシンキナーゼ阻害薬であるトレブルチニブは自然免疫系にも作用し神経変性にも有効な可能性がある。
カンナビノイド
海外では、カンナビノイドはナビキシモルス(商標名サティベックス:Sativex)として神経因性疼痛、痙縮、過活動膀胱、ほかの症状の緩和に用いられている。
日本ではカナビスの主要活性成分であるテトラヒドロカンナビノールが進行を遅くすることを示唆する研究結果が得られたため、臨床試験が行われている。
免疫グロブリン療法
トピックス
進行型多発性硬化症
一次進行型多発性硬化症(PPMS)と二次進行型多発性硬化症(SPMS)は同じentityと考えられており両者をあわせて進行形MS(progressive MS、PMS)という。ゲノムワイド関連解析ではPPMSと他の病型で遺伝学的な明確な差異が認められなかった。またPPMSとSPMSを明確に鑑別できるバイオマーカーが確立していない。病理学上、白質の脱随斑の広がり、病変のサブタイプ(活動性/慢性活動性/非活動性病変)の発生率に差がない。皮質病変もSPMSとPPMSで発生率に大きな差がない。これらを根拠にPPMSとSPMSは同じentityと考えられている。PMSでは無症候性のMRI上の造影病変ないし新規・拡大T2延長病変が出現する場合があることが知られている。この活動性のあるactiveなグループを分けて考えることの方がSPMSやPPMSといった従来の分け方より重要と考えられている。
慢性炎症と神経変性
多発性硬化症は中枢神経系の脱髄性疾患でありその背景には2つの病態が存在することが知られている。一つは免疫介在性のメカニズムによる慢性炎症ともうひとつは神経変性である。多発性硬化症は炎症性疾患とされることが多いが、初期には炎症性病態が主に働いているものの、実は変性過程も病初期から存在し、疾患の経過とともに炎症性病態の占める割合が低下し、それにつれ変性過程が主になってくると考えられている。多発性硬化症の炎症性病変には2つのタイプがある。ひとつは血液脳関門の破綻により大量のT細胞とB細胞が脳内に流入し、活動性の脱髄病変をきたすものであり典型的な病理としてはCD8陽性細胞、CD20陽性細胞、少数の形質細胞が中心静脈周囲に炎症性細胞浸潤を形成し、マクロファージが大量に浸潤する。もうひとつのタイプは血液脳関門の破綻がないにもかかわらず、血管周囲やVirchow-Robin腔や髄膜の結合組織にT細胞やB細胞がゆっくり集積していくものである。前者は急性期多発性硬化症でみられるものでどちらかというとRRMSに特徴的なパターンであり、後者は初期にすでに認められるが罹病期間の長さや患者の年齢があがるにつれ徐々に増加していく、どちらかというとPMSに特徴的なパターンである。またRRMSでは少なからぬリンパ球浸潤が認められるのに対してPMSではミクログリア/マクロファージの介在する炎症が優勢になるという違いも存在する。
病態
PMSに特徴的な病理所見としてまず緩徐拡大病変(slow expanding lesion、SEL)があげられる。これは再発もなく、MRI上で造影病変や新規ないし拡大T2延長病変といった活動性病変も認められないのに障害が進行する、smouldering MSあるいはprogression independent of relapse activity(PIRA)の原因の一因となっていると認知されてきた病変である。この病変は非常にゆっくり拡大していき、病理学的にはCD8陽性T細胞、CD20陽性B細胞、形質細胞などで構成された炎症性浸潤からのなんらかの液性因子出て辺縁にある活性化したミクログリア、その部位でみられる活発な脱髄から構成される。このため炎症性浸潤からなんらかの液性因子が出て、辺縁のミクログリアが活性化されていると想定されている。ミクログリアの活性化を介して脱髄と神経変性が誘導されると考えられている。
ミクログリアを活性化させる液性因子は明らかにはなっていないが、セマフォリンやセラミドなどが候補として挙げられている。辺縁の活性化したミクログリアはしばしば鉄を含んでおり、鉄を含むSELは拡大のスピードが速く、この病変を多く認める患者は認知機能障害や運動障害が若くして認められるなどのデータからこの鉄蓄積の病態への関与が疑われている。その他の病理学的特徴として髄膜の主にB細胞と形質細胞からなる白血球集積があり、これはときに三次リンパ濾胞様構造を成し、皮質性の脱髄に寄与している。
これらの病理学的基盤となる病態としてミトコンドリア機能異常が知られている。MS病変においてミトコンドリア呼吸鎖複合体Ⅳの異常が報告されている。ミトコンドリア機能異常のさらなる原因として酸化ストレスの関与が疑われている。SEL病変で述べた鉄沈着は、炎症後にヘモグロビンの変性に伴い生じると考えられており深部灰白質や骨髄系細胞周囲にも認められており、活性酸素の産出を促進し、炎症性サイトカイン産出にむすびつくのではないかと考えられている。
脚注
- 難病情報センター - 特定疾患情報、多発性硬化症
- 国立精神・神経医療研究センター - 多発性硬化症センター(MSセンター)
- 多発性硬化症 (MS) MSDマニュアル プロフェッショナル版
参考文献
- 最新アプローチ 多発性硬化症と視神経脊髄炎 ISBN 9784521734415
- 多発性硬化症(MS)診療のすべて ISBN 9784787818188
- 多発性硬化症と視神経脊髄炎の基礎と臨床 ISBN 9784753225767
関連項目
外部リンク
- 国立精神・神経医療研究センター 神経研究所 免疫研究部 - 研究情報
- 東北大学多発性硬化症治療学寄附講座 - 研究情報
- 宇多野病院 - 多発性硬化症センター (MSセンター)
- 多発性硬化症 - 脳科学辞典
- Multiple Sclerosis - ウェイバックマシン(2012年10月12日アーカイブ分) (英語) Medpedia「多発性硬化症」の項目。
- カナビススタディハウス - アメリカなどにおける医療カナビス(大麻)法の対応疾患
- YouTube - アメリカにおける医療大麻の実態
- 山村隆、「腸内細菌と多発性硬化症」『日本内科学会雑誌』 2016年 105巻 9号 p.1717-1721, doi:10.2169/naika.105.1717, 日本内科学会